Choose language
Choose language
< Return to main menu
Choose language



News & Events

Services & Solutions
News & Events
Career
Platform
Magical Power of Quantum Mechanics
Events
News
Magical Power of Quantum Mechanics
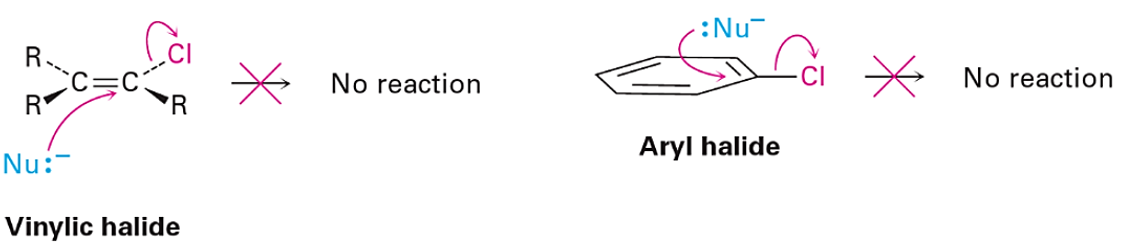
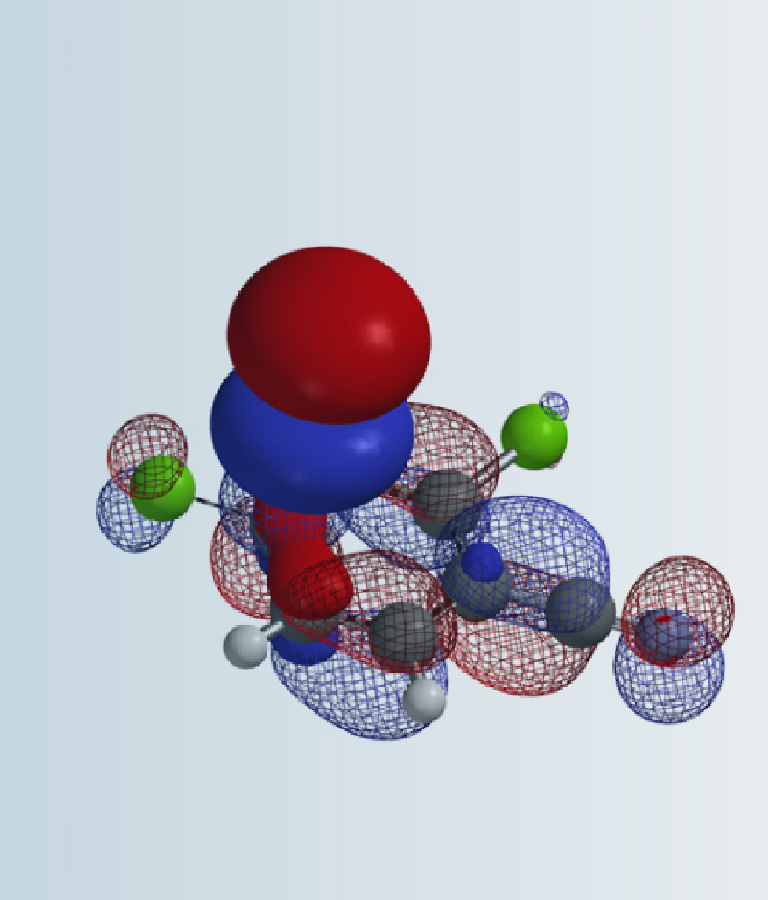
Alkyl and vinyl halides are important reagents and intermediates in organic synthesis. Shown in figure 1 are LUMO and LUMO+1 of ethyl and vinyl chlorides. It is noteworthy that LUMO of ethyl chloride is very similar to LUMO+1 of vinyl chloride, suggesting that they could react in similar manners. This is consistent with the observations that both of them react with magnesium to form the corresponding Grignard reagents, and undergo dehydrohalogenation to generate ethylene or acetylene. With ethyl chloride, interaction of the LUMO lobe on the C-Cl carbon with HOMO of incoming nucleophile leads to nucleophilic substitution reaction at the sp3 carbon with inversion of configuration (SN2 reaction). Intuitively, this leads to the question, can vinyl chloride undergo a similar in-plane SN2 reaction?
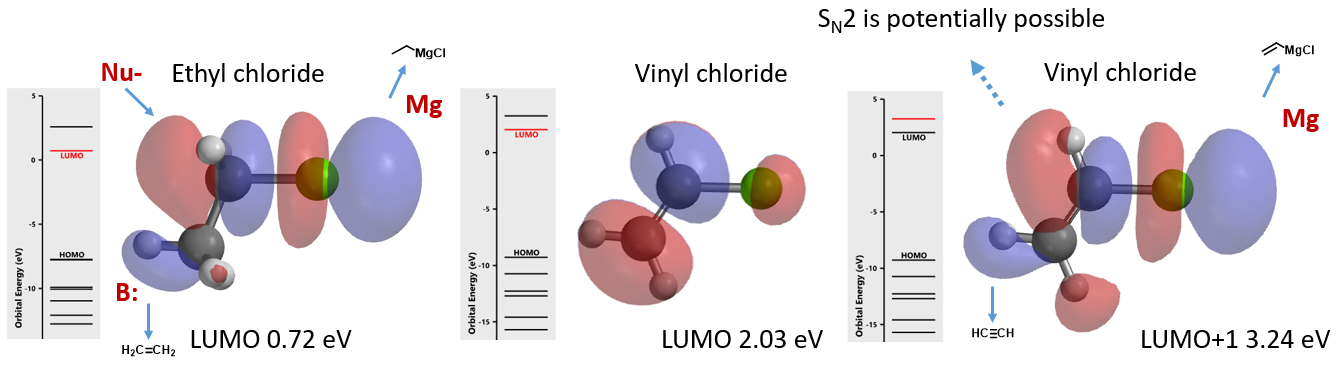
Figure 1. LUMO of ethyl chloride and LUMO, LUMO+1 of vinyl chloride
In 1994, Pross and Radom et al. predicted by ab initio calculations that in-plane SN2 reaction with configuration inversion of unactivated vinyl chloride is possible in both gas phase and solution[1]. In 2004, Ando and Narasaka et al. demonstrated the first in-plane SN2 reaction of vinyl chloride E-1 with DFT calculations, then confirmed it experimentally (Figure 2)[2]. Substrate is configuration specific for the reaction. When E-1 is treated with NaH in DMF solvent, the reaction proceeds at room temperature to provide benzofuran 2 in 95% isolated yield. On the other hand, the isomeric Z-1 was recovered quantitatively after heating at 110 °C.
For calculation of the transition state (Figure 2), Ando and Narasaka et al. used the B3LYP/6-31+G* (Onsager continuum, DMF e=37) model. Since the exact coordinate data was not reported, we repeated the calculation with wB97X-D DFT. The calculated transition state has a carbon-oxygen distance of 2.158 Å, close to the value reported, and an imaginary frequency of i556 cm-1, corresponding to the bonds being made and broken (Figure 3).
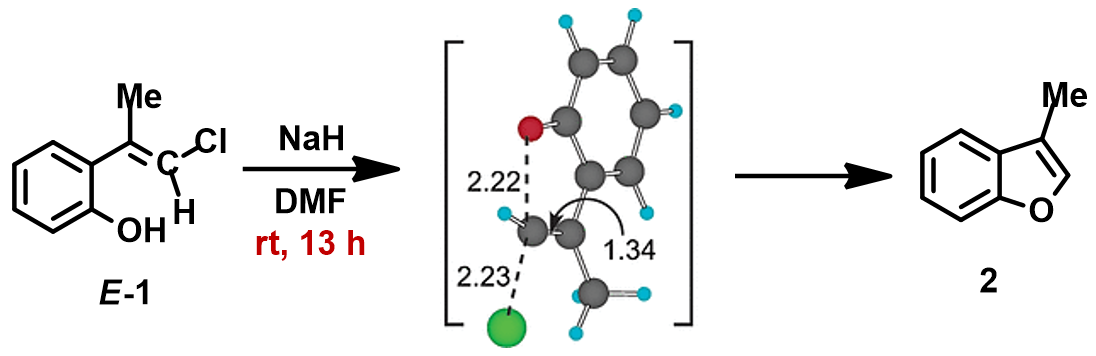
Figure 2. E-1 Intramolecular SN2 reaction and reported transition state structure
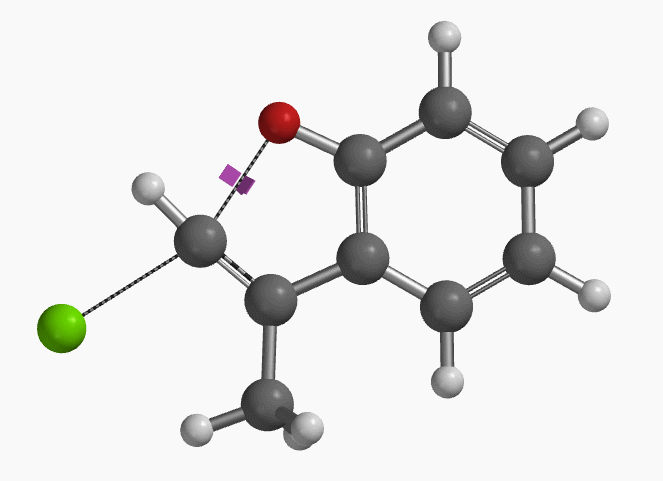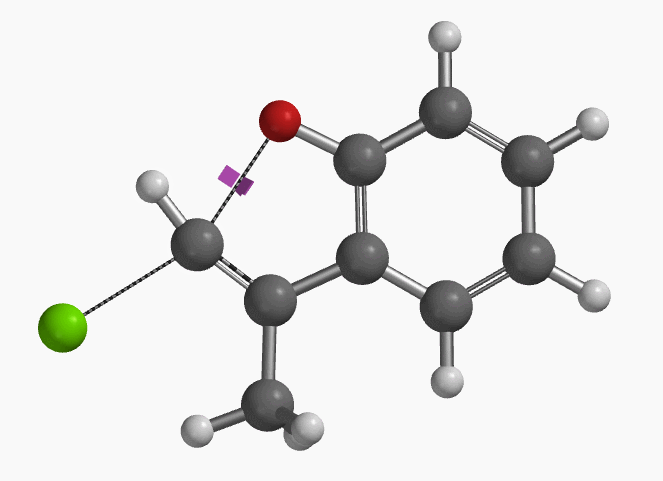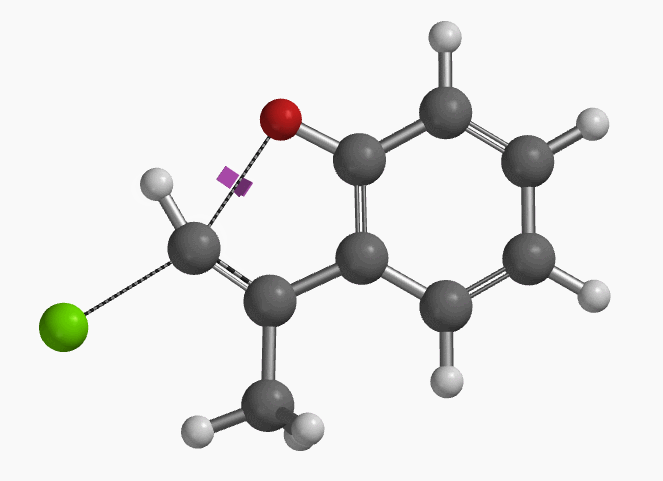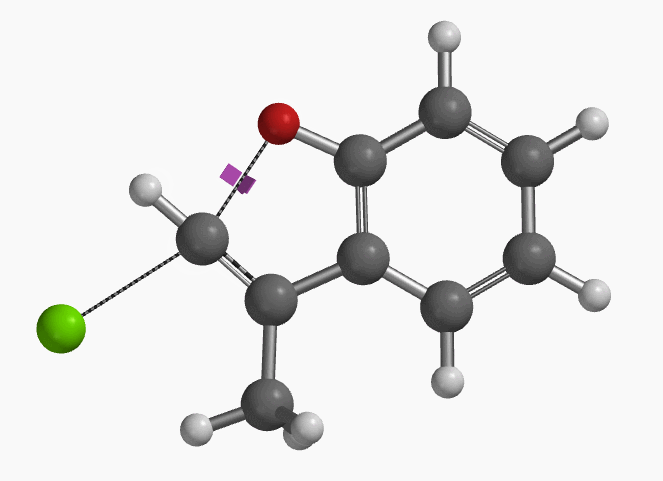
Figure 3. Transition state structure of E-1 SN2 reaction with an imaginary frequency of i556 cm-1
With the coordinate of transition state, we calculated for the intrinsic reaction coordinate (IRC)[3] of the reaction (Figure 4). This shows that as the oxyanion approaches the sp2 carbon, the vinylic hydrogen gradually flips from the inner side to the outer side, in the plane defined by Cl, C, O atoms, while the chloro group is being displaced. The carbon-chlorine bond breaks and the carbon-oxygen bond forms in a concerted manner, analogous to the umbrella-like inversion at sp3 carbon in SN2 reaction.
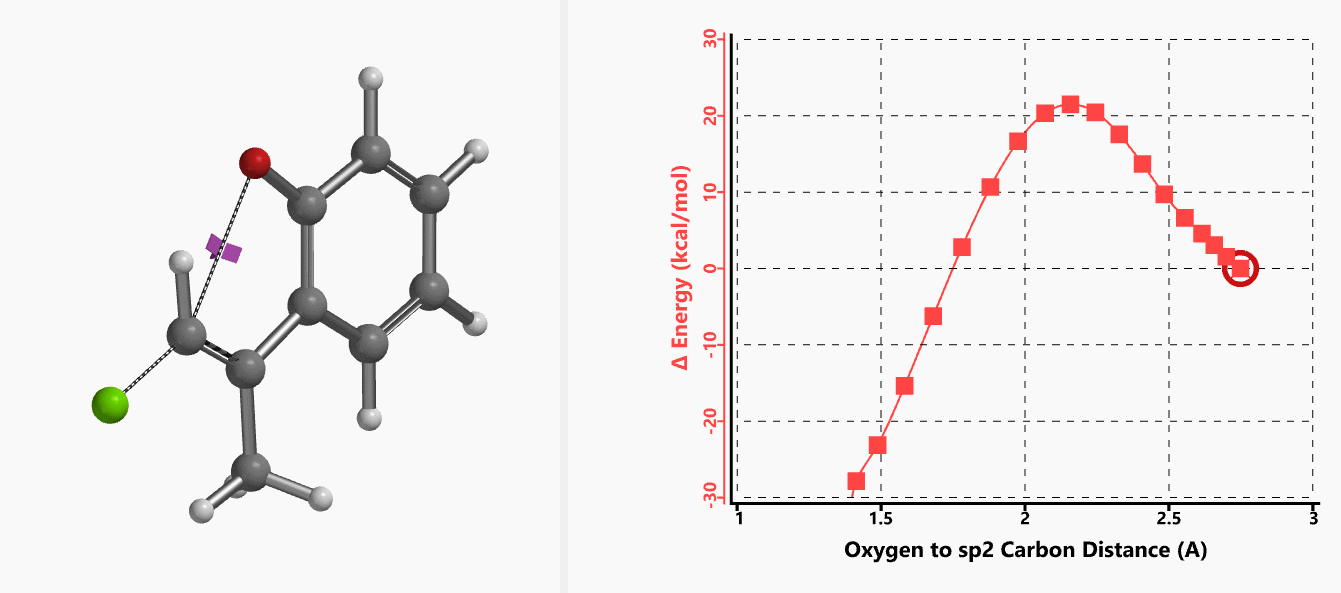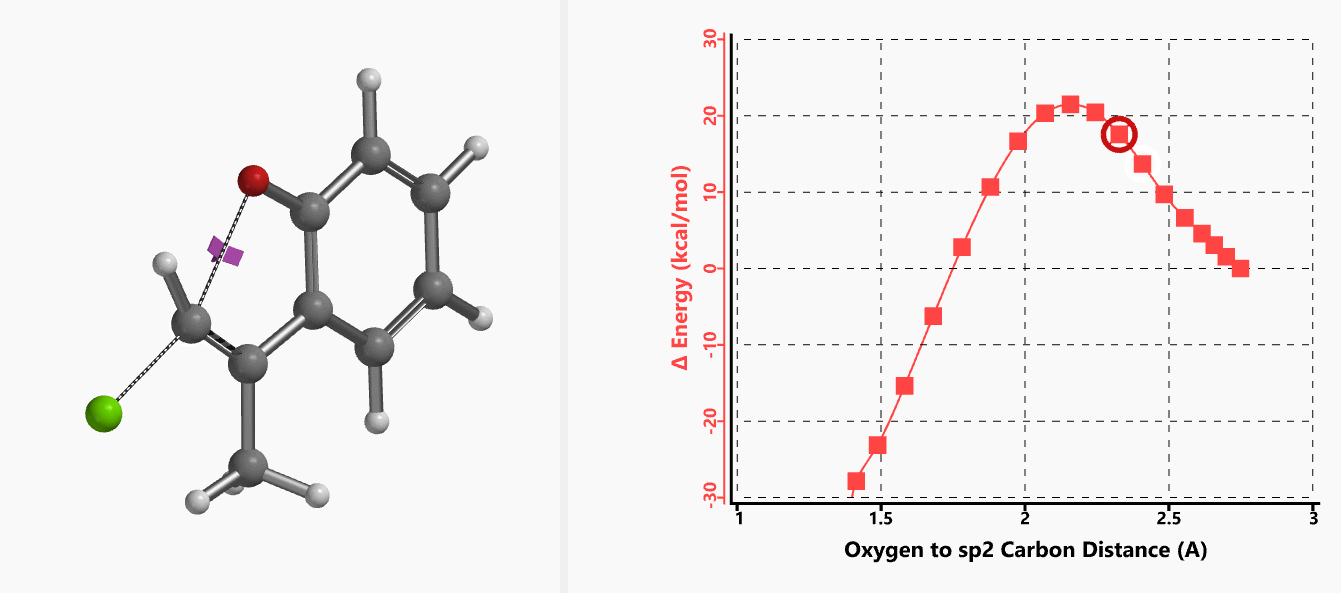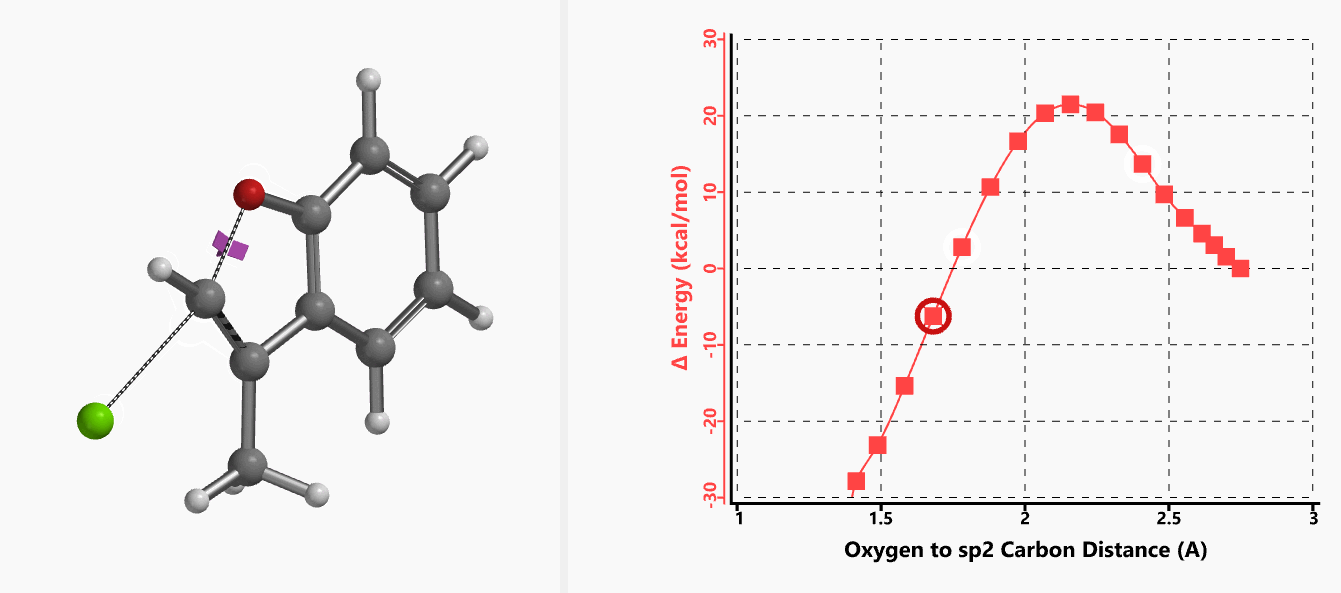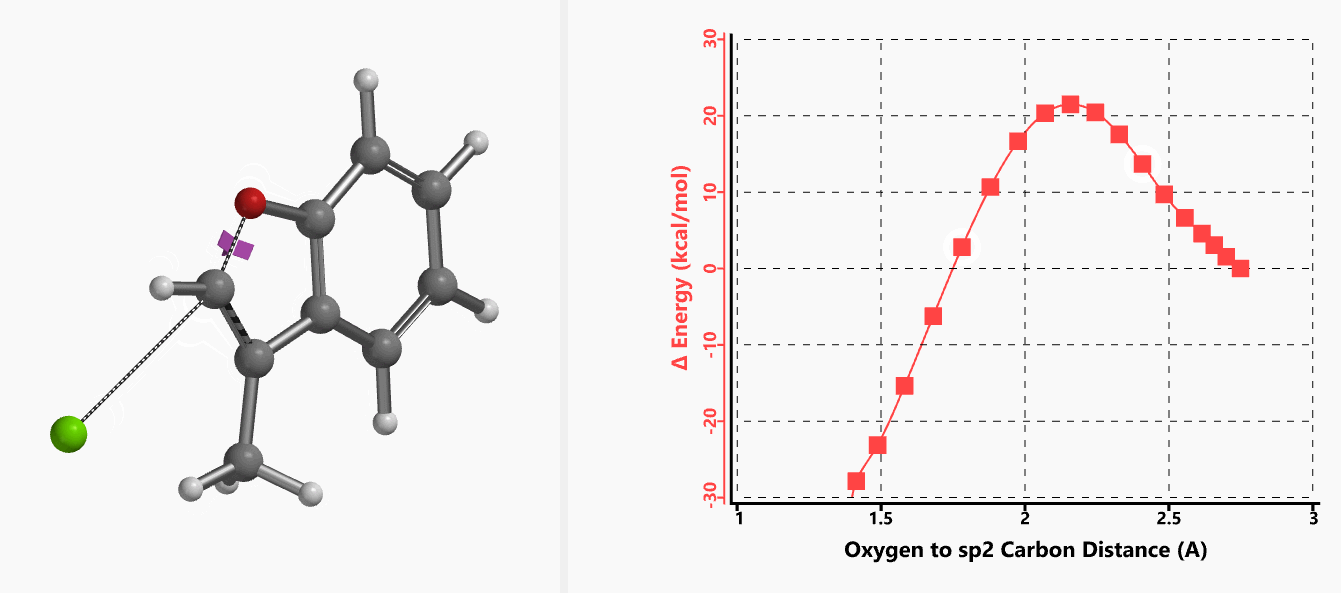
Figure 4. Intrinsic reaction coordinate of E-1 sp2 SN2 reaction
Shown in Figure 5 are changes in electron density contour across the plane of the molecule as reaction proceeds. Electron density between the oxyanion and the vinyl hydrogen gradually increases toward the transition state, indicative of hydrogen bond interaction between them. When the distance between the oxygen and sp2 carbon is approximately 2.4 Å, the hydrogen atom begins to flip to the other side. The electron density gradually shifts from between oxyanion and vinyl hydrogen to between oxygen and sp2 carbon. These account for the remarkable sp2 SN2 reactivity observed with the E-1 isomer.
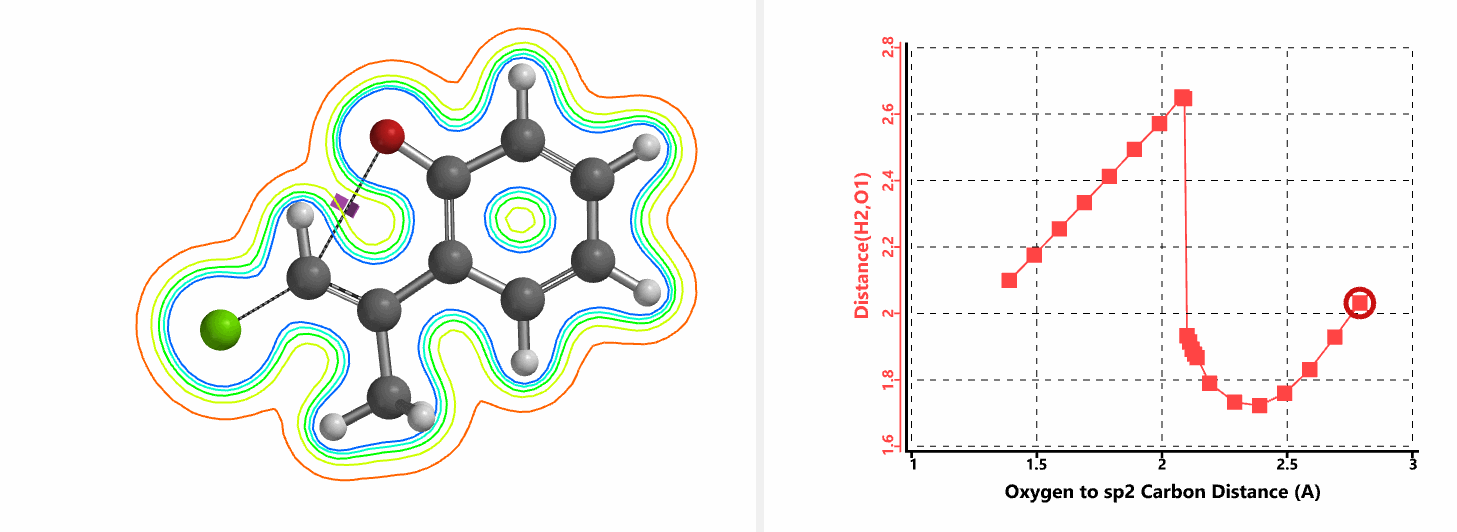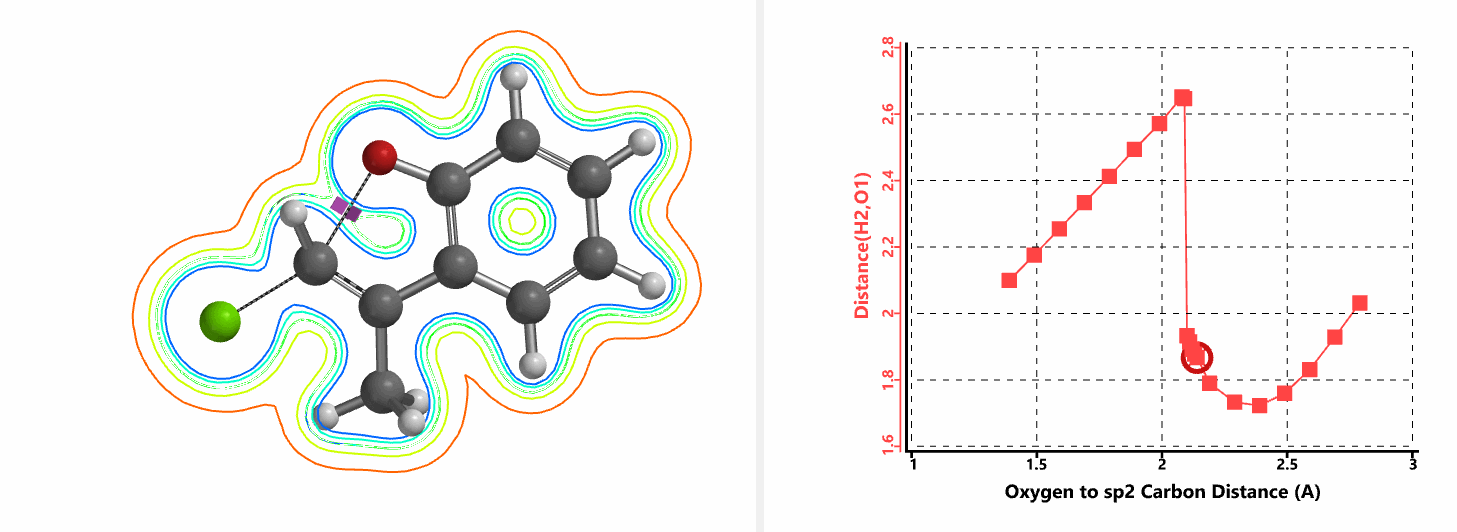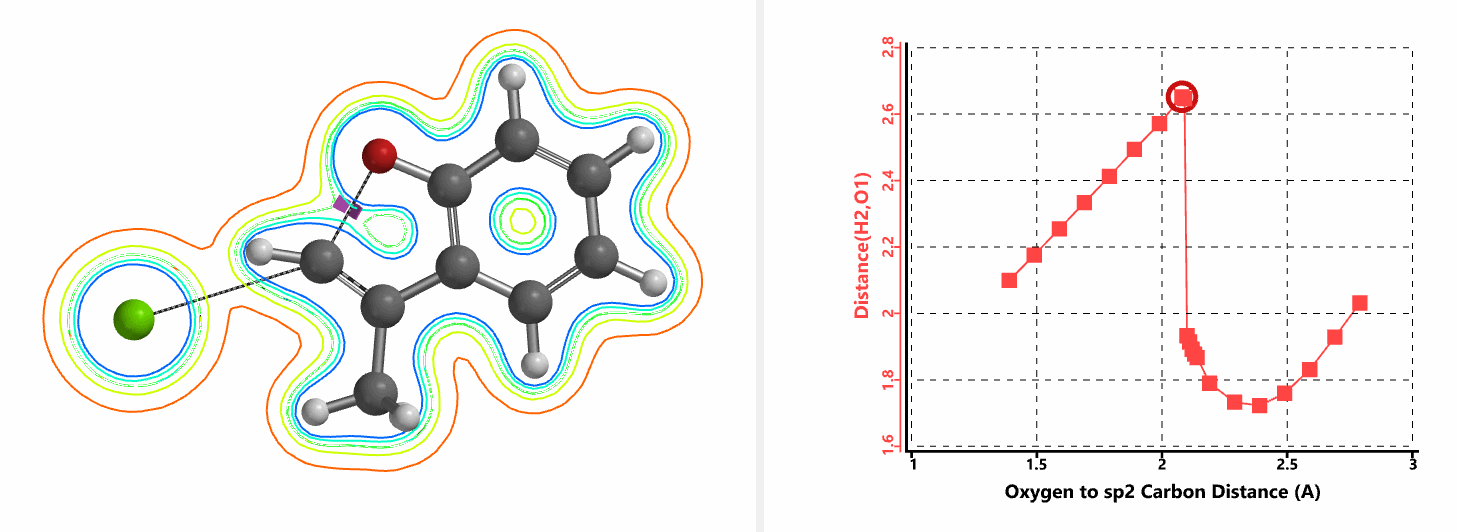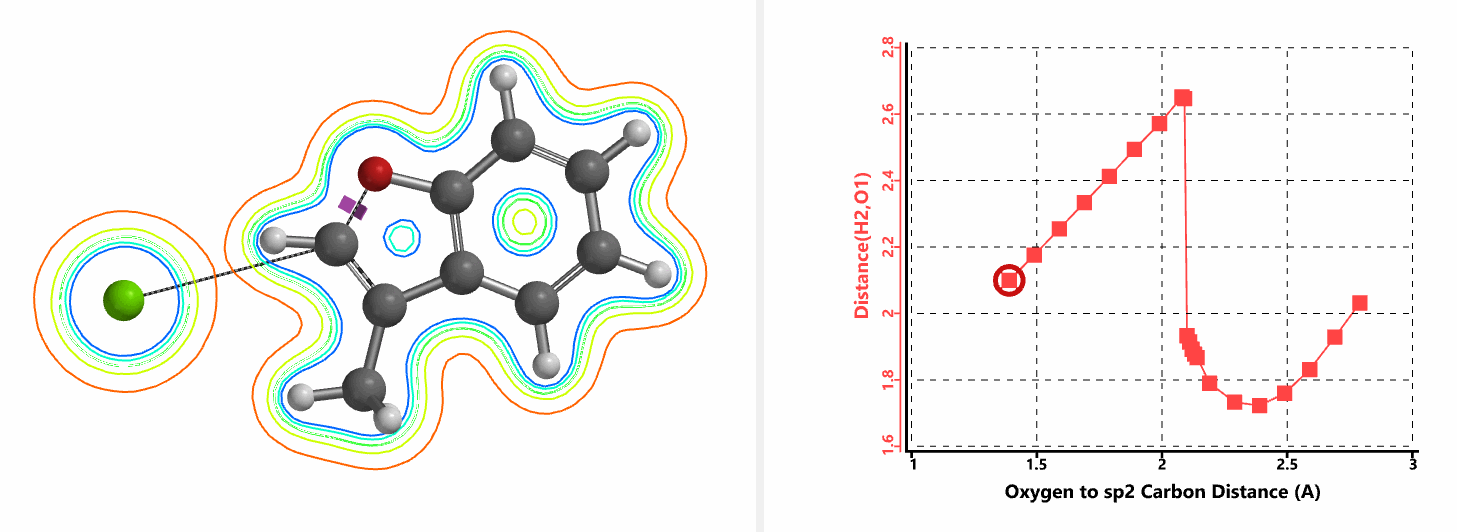
Figure 5. Changes in electron density contour as E-1 sp2 SN2 reaction proceeds
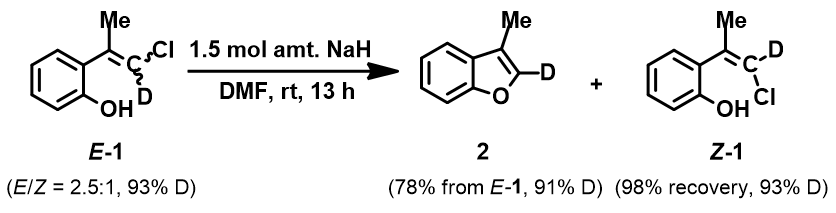
Figure 6. Reaction with a mixture of deuterium labeled E-1 and Z-1
When a mixture of E-1 and Z-1 deuterium labeled substrate was treated with NaH in DMF at room temperature, E-1 was consumed, benzofuran 2 with deuterium label at C-2 was isolated, while Z-1 was almost quantitatively recovered (Figure 6). These results provide further support for the sp2 SN2 mechanism discovered with DFT calculations, and exclude possibility of the alternative allylic isomerization and carbene insertion mechanisms (Figure 7).[4]
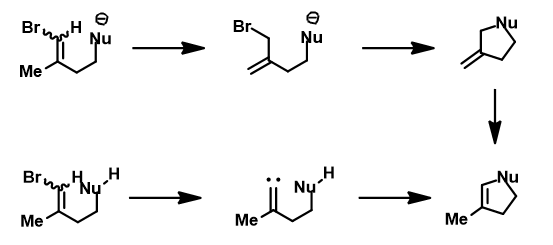
Figure 7. Excluded possible allylic isomerization and carbene insertion mechanism
Further examples of in plane sp2 SN2 reactions were reported, along with computational studies.[4]
Figure 8 summarizes substrate scope for sp2 SN2 reaction with carbon reaction center:
1.Nucleophile could be oxygen, nitrogen, carbon, or sulfur based[4]
2.Leaving group X could be Cl, Br
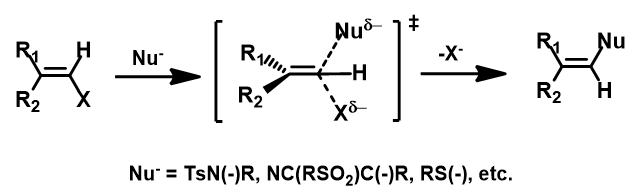
Figure 8. Substrate scope for sp2 SN2 reaction with carbon reaction center
In 2020, Chen et al. reported an efficient method for the synthesis of 2-aryl-1,2,3-triazoles via intramolecular nitrogen-nitrogen bond formation (Figure 9)[5]. Selective activation of dimethyl(phenylhydrazono)ethylidenehydrazines with methyl iodide provided hydrazonium intermediates 3, which readily cyclized in the presence of base to provide a variety of 2-aryl-1,2,3-triazoles 5 in good to excellent yields. The authors suggest that the reaction proceeds via 4b, with an intramolecular SN2 on the sp2 nitrogen, displacement of trimethylamine, and formation of a new N–N bond. This is substantiated with DFT computational studies.
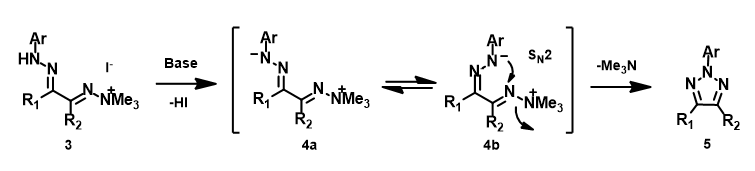
Figure 9. Proposed intramolecular SN2 reaction mechanism for synthesis of 2-aryl-1,2,3-triazoles
Intrigued by their findings, we calculated the transition state (Figure 10, iFreq i458 cm−1) and then intrinsic reaction coordinate (IRC) of the reaction (Figure 11). The latter illustrated in detail the in-plane sp2 nitrogen SN2 concerted process, with relatively lower energy barrier than the sp2 vinyl halide SN2 reaction described above, presumably due to the absence of the vinylic hydrogen. This reaction is an excellent application of the sp2 SN2 mechanism. Arylation of triazole with Chan-Lam reaction will provide a mixture of N-1 and N-2 arylation products in low yield[6].
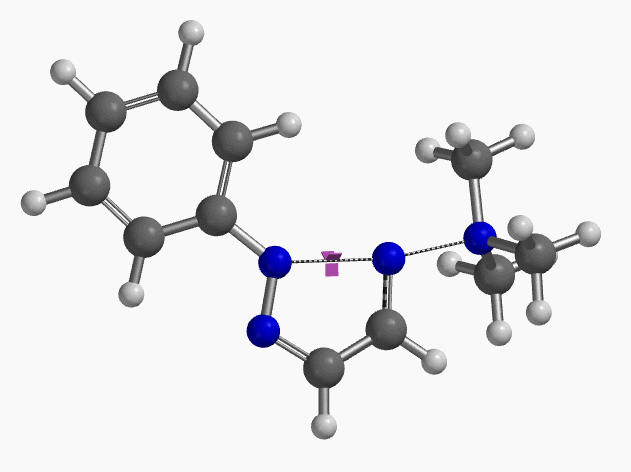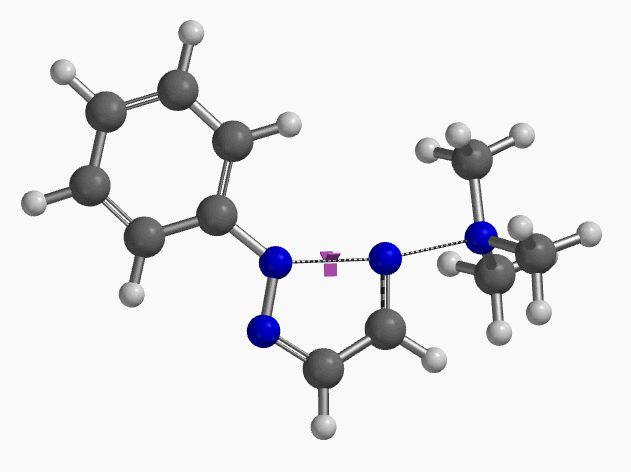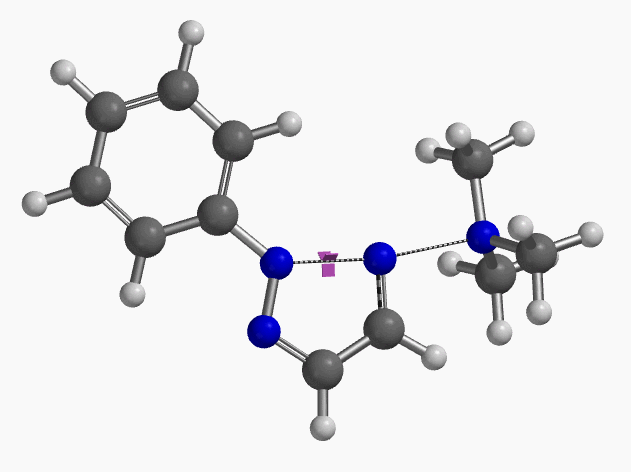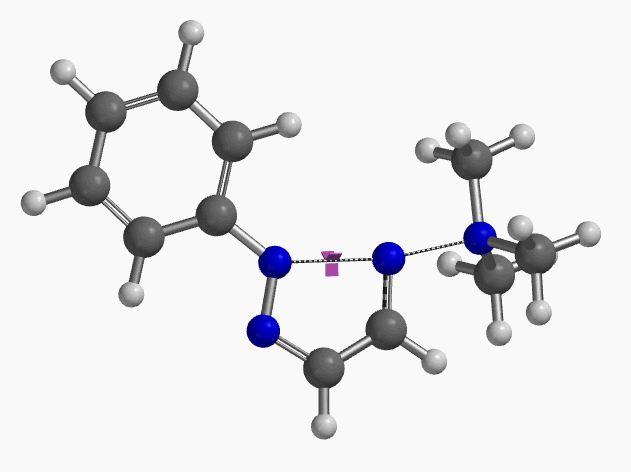
Figure 10. Transition state, with imaginary frequency of i458 cm-1, in the formation of 2-aryl-1,2,3-triazole
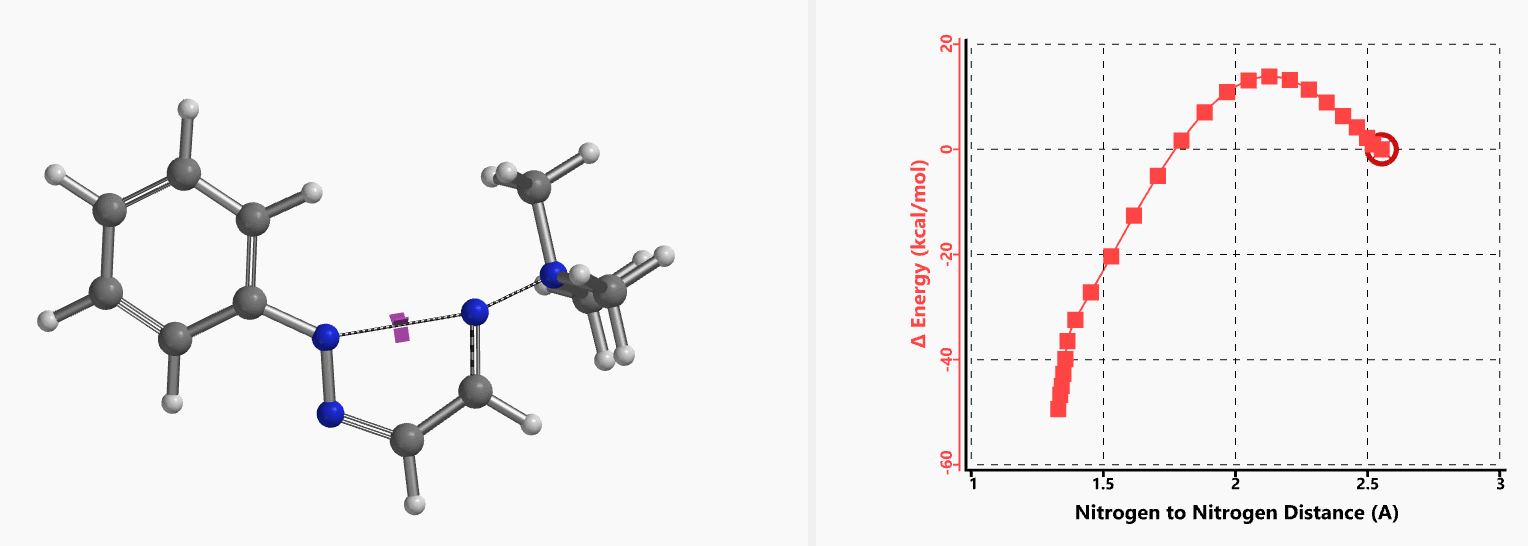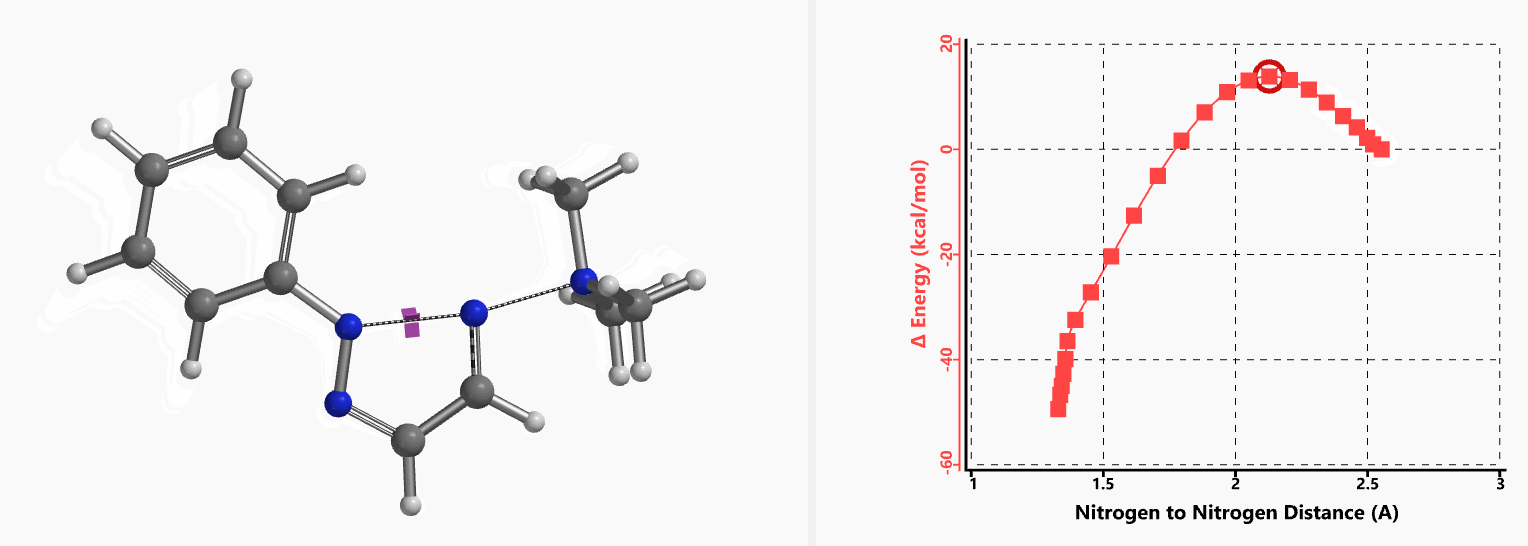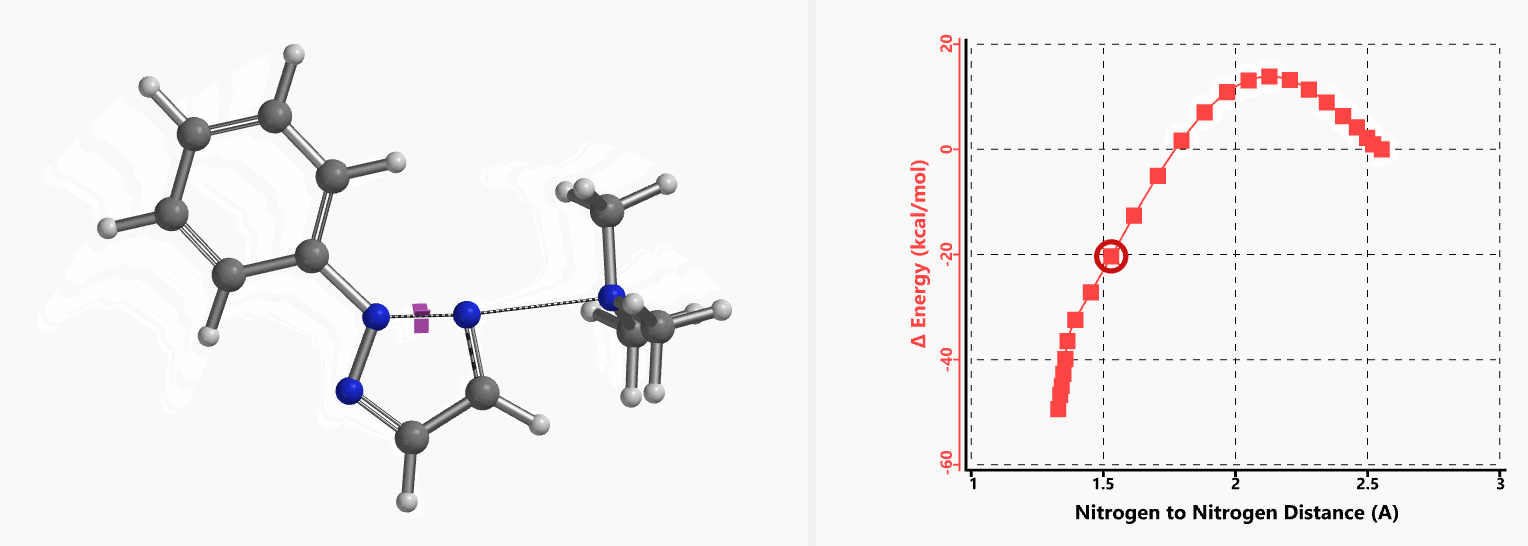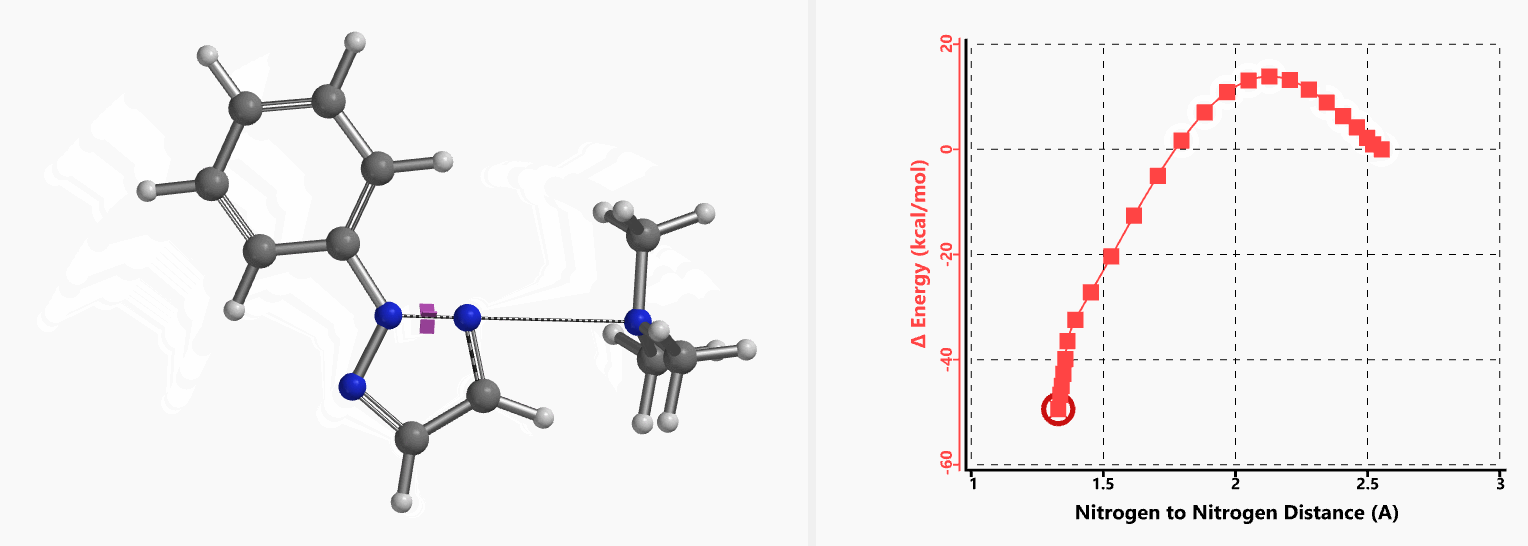
Figure 11. Intrinsic reaction coordinate for formation of 2-aryl-1,2,3-triazole
In-plane sp2 SN2 reactions are considered unlikely on the basis of the perception that SN2 attack with inversion at vinylic centers is a high-energy process[1,7]. Quantum mechanics give organic chemists data and insight to objectively reassess the feasibility of such reactions [1, 2]. In this chapter we learned:
1) SN2 reactions at sp2 carbon and nitrogen (SNVσ) can occur and are predictable
2) Accessibility of the sp2 reaction center to nucleophilic attack determines feasibility of such sp2 SN2 reaction
3) Potential application/occurrence of these sp2 SN2 reactions in retrosynthetic planning[4,5]
4) Quantitative QM analysis can provide us with excellent guidance on their feasibilities.
Based on the LUMO, LUMO/Electron Density overlay of chlorobenzene (Figure 12), a nucleophilic substitution reaction of it via a sp2 SN2 mechanism will be sterically impossible, would you agree?
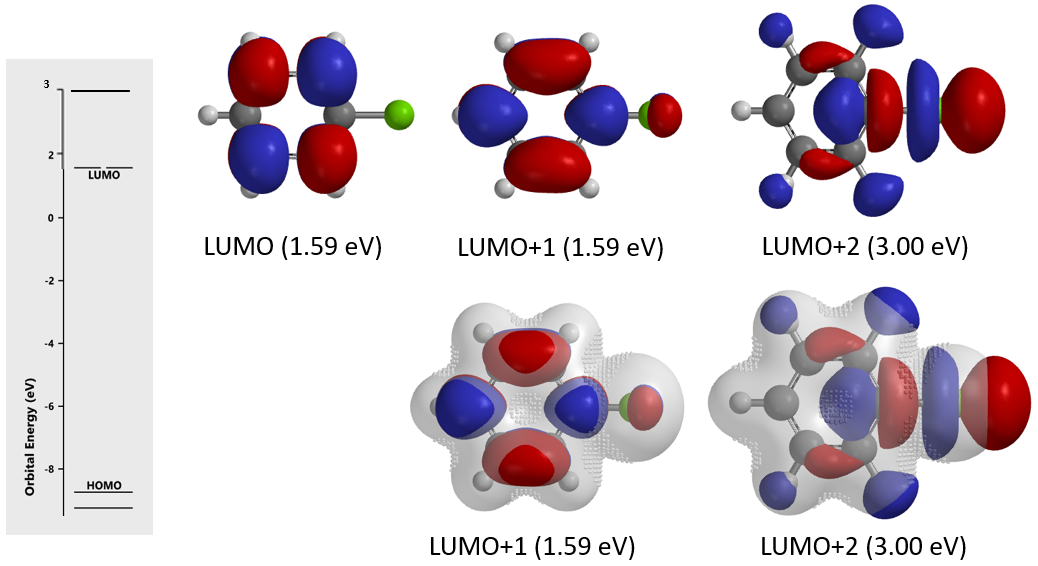
Figure 12. LUMO and LUMO/Electron Density overlay (with inaccessibility markers) of chlorobenzene
This article is written and edited by Liting Dong, Tommy Lai, Yongsheng Chen, and John S. Wai.
References:
[1] M. N. Glukhovtsev, A. Pross, L. Radom J. Am. Chem. Soc., 1994, 116, 5961. In-plane SN2 was considered unlikely on the basis of the perception that SN2 attack with inversion at vinylic centers is a high-energy pathway than out-of-plane p-attack. Standard ab initio molecular orbital calculations showed that in-plane σ-type SN2 substitution for Cl- + CH2=CHCl with inversion at unactivated sp2 carbon (136.5 kJ/mol), is actually energetically preferred to the out-of-plane p-pathway (178.9 kJ/mol) by 42.4 kJ/mol.
[2] K. Ando, M. Kitamura, K. Miura, K. Narasaka Org. Lett., 2004, 6, 2461.
[3] QM Organic Chemistry Classroom Chapter 28 "A QM Study of the Hydroaminomethylation of Olefins".
[4] H. Miyauchi, S. Chiba, K. Fukamizu, K. Andob, K. Narasaka, Tetrahedron, 2007, 63, 5940.
[5] C.Y. Chen, X. Lu, M. C. Holland, S. Lv, X. Ji, W. Liu, J. Liu, D. Depre, P. Westerduin, Eur. J. Org. Chem., 2020, 548.
[6] QM Organic Chemistry Classroom Chapter 34 "QM Analyses of Regioselectivity in Chan-Lam Reaction".
[7] J. McMurry, Organic Chemistry, Eighth Edition, Boston, MA, USA: Cengage Learning, 2011; pp 379-380. “Vinylic halides and aryl halides are….unreactive towards SN2 displacement. This lack of reactivity is due to steric factors: the incoming nucleophile would have to approach in the plane of the carbon-carbon double bond and burrow through part of the molecule to carry out a backside displacement.”