 Choose language
Choose language
< Return to main menu
Choose language



News & Events

Services & Solutions
News & Events
Career
Platform
Magical Power of Quantum Mechanics
Events
News
Magical Power of Quantum Mechanics
We discussed the use of QM calculated activation energy to analyze regio-selectivity alkylation of pyrazole in Chapter 9. In this chapter we’ll discuss its application on SNAr reactions of polyhalogenated heterocyclic compounds.
For the dichloropyrazolopyrimidine carboxylate 1 in Figure 1, there are potential three sites for nucleophilic attack, C1, C2 on the pyrimidine ring, and C3 on the ester moiety. What will happen when we treat compound 1 with aqueous sodium hydroxide in tetrahydrofuran? Is there a reliable way for predicting reaction outcome?
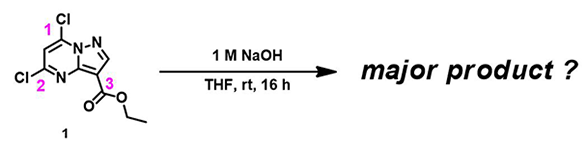
Figure 1. Hydrolysis of ethyl 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carboxylate 1
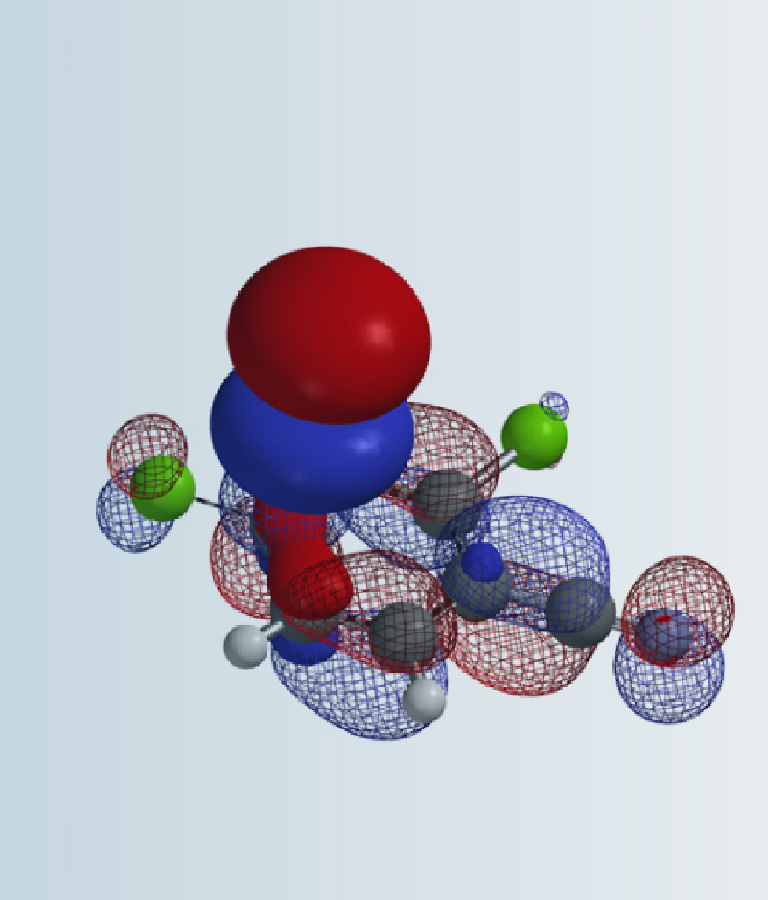
Since compound 1 is an electrophile, we first look at its LUMO (Figure 2).
There is close to none orbital lobe on C3, nucleophilic attack of the ester group is not likely to happen. There are significant LUMO lobes on C1 and C2, however they are similar in size, and their accessibility could not be differentiated with the calculated LUMO Map (Figure 2). Under these circumstances, we will proceed to calculate and compare the activation energy difference of these two potentially competing nucleophilic reactions.
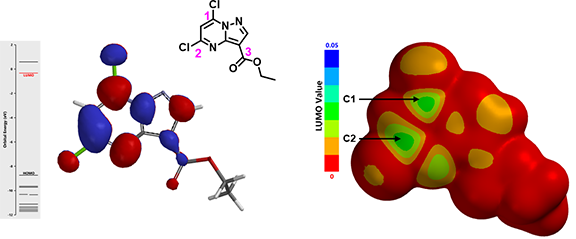
Figure 2. LUMO (left) and LUMO Map (right) of compound 1
We learned from experience that using bromide as a surrogate nucleophile to model this kind of reaction could provide a more reliable estimate. This will avoid overestimation of hydrogen bonding effect occasionally observed between hydroxide anion and relatively electron-deficient hydrogen on the aromatic system.
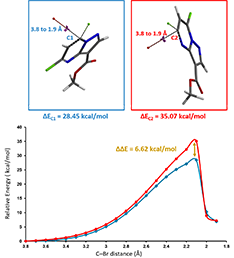
Figure 3. Activation energies of competing SN2 reactions at C1 (blue) and C2 (red)
Shown on Figure 3 are the two reaction energy profiles calculated. Activation energies required for reactions at C1 and C2 are 28.45 and 35.07 kcal/mol, respectively, a difference in 6.62 kcal/mol, in favor of nucleophilic attack at C1. Referring to Table 1 (discussed in Chapter 9), it becomes obvious that the hydrolysis will be highly selective at C1.
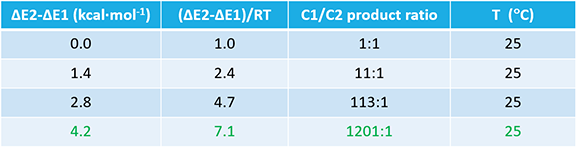
Table 1. Product ratio obtained with different ΔΔE and temperature
After we treated compound 1 with aqueous sodium hydroxide in tetrahydrofuran at room temperature, we saw preferentially reaction at C1 position to provide compound 2, the only product [1, 2] (Figure 4). Neither nucleophilic substitution of the C2 chloro group nor hydrolysis of the ester group was observed.
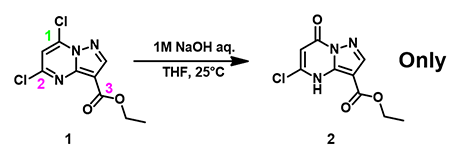
Figure 4. Selective hydrolysis of 5,7-dichloropyrazolo[1,5-a]pyrimidinecarboxylate 1
In summary, for QM analysis of SNAr reaction, we first calculate LUMO and LUMO Map of the substrate to look for differences in LUMO lobe and their accessibility for nucleophilic interactions. When further differentiation is needed or an estimation of the product ratio is required, we will calculate for and compare the activation energies of the competing reaction paths.
Building on What We Just Learn
For the chloropyridines below, they have a unique order of reactivity [3].

Figure 5. 4-chloro (A), 2-chloro (B), and 3-chloropyridines (C)
The relative differences in calculated activation energies correlate well with the reactivity pattern observed.
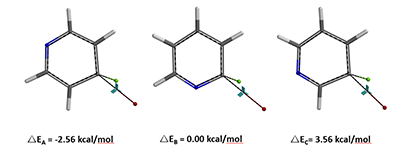
Figure 6. Relative differences in calculated activation energies of SNAr reaction on 4-, 2-, and 3-chloropyridines
Then we arrive at an intriguing question. Why 4-chloropyridine is more reactive than 2-chloropyridine? The aromatic nitrogen is further away in the 4-chloro analog [4].
This article is written and edited by Xiaocang Wei, Qiuyue Wang, Guqin Shi, and John S. Wai.
References:
[1] R. K. Robins, G. R. Revankar, et al. J. Heterocycl. Chem. 1985, 22, 601
[2] J. R. Zbieg, P. P. Beroza, J. J. Crawford, WO2019232216
[3] J. A. Joule & K. Mills, Heterocyclic Chemistry, 5th edition, 2010, p117-118
[4] Hint: Compare and contrast the LUMO and LUMO+1 of the chloropyridines.