 Choose language
Choose language
< Return to main menu
Choose language



News & Events

Services & Solutions
News & Events
Career
Platform
Magical Power of Quantum Mechanics
Events
News
Magical Power of Quantum Mechanics
Dimethyl carbonate (DMC) is a low toxicity, fast biodegradability chemical feedstock and important organic reagent. DMC, structurally consisting of a carbonyl and two methoxy functional groups, is an excellent substitute for highly toxic phosgene and dimethyl sulfate in acylation and methylation, respectively.
Hard-Soft-Acid-Base (HSAB) principle was introduced by Pearson in 1963 [1] and is being widely used to explain stability of reaction products and mechanisms, where "hard" refers to higher charge density and smaller radius species (ions, atoms, molecules) that are weakly polarizable with greater polarity, while "soft" means lower charge density and larger radius species that are strongly polarizable with lower polarity. HSAB principle qualitatively explains reactivity patterns with "hard likes hard, soft likes soft" preference.
In 2005, Tundo et al. published their observations on how ambident DMC reacts differently with phenols, thiophenol, and aniline under various reaction conditions [2]. However, their application of the HSAB principle failed to explain the experimental results in a consistent manner, creating more confusion. In 2011, Mayr et al. advocated "Farewell to the HSAB Treatment of Ambident Reactivity" [3] and revealed that thermodynamic/kinetic control could better account for reactivity of organic ambident reagents observed (Figure 1).
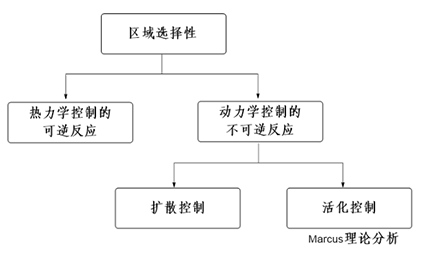
Figure 1: A systematic approach to ambident reactivity [3]
Inspired by Mayr et al., we found that correlating the LUMO & LUMO+1 characteristics of DMC and pKa values of the substrates (phenols, thiophenol, and aniline) is indeed sufficient to understand the complex ambident reactivity patterns of DMC, avoiding the unnecessary confusion with HSAB analysis [4].
As an ambident electrophilic reagent, DMC can react differently with nucleophilic reagents to produce the carboxymethylation or methylation product or both (Figure 2). When the carbonyl carbon of DMC is attacked by the nucleophile, the acyl-oxygen bond is broken, forming an acylation product, accompanied by the formation of CH3O-; when the methoxy carbon of DMC is attacked by the nucleophile, the methyl-oxygen bond is broken, forming a methylation product, accompanied by the formation of CH3O- and CO2. Below is our analysis of DMC reactivity patterns with various phenols, thiophenol, and aniline.
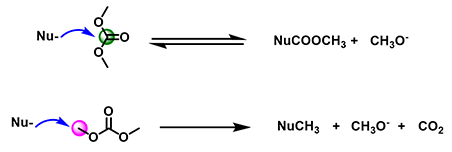
Figure 2: Ambident reactivity of Dimethyl Carbonate
Shown on Table 1 are the results reported by Tundo et al. on how various phenols react with DMC to give different relative percentage of methylation vs. carboxymethylation products, along with pKa values of various phenols we tabulated from the Reich and Bordwell pKa compilation [5]. For p-methoxyphenol, acylation product is predominant; while for p-cyanophenol, only the methylation product is formed.

Table 1: Reaction of phenols with different substituents with DMC [2]
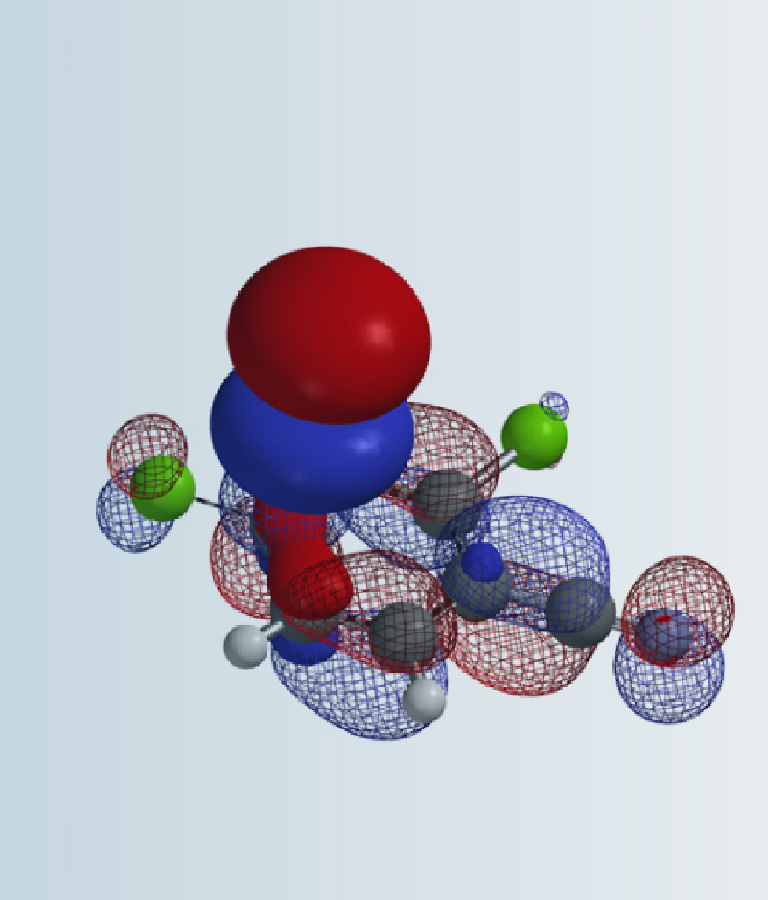
Since DMC is an electrophile, we calculated for its LUMO and LUMO Map first [6]. See our Chapter 1 for LUMO analysis of electrophiles.
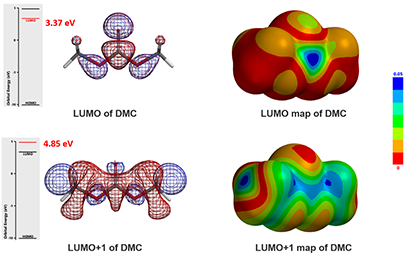
Figure 3: DMC's LUMO/LUMO + 1 and LUMO/LUMO + 1 map
LUMO (3.37 eV) and LUMO map of DMC show accessible reaction site on carbonyl carbon, while LUMO + 1 (4.85 eV) and LUMO+1 map show the accessible reaction site on methoxy carbon (Figure 3), with a significant orbital energy difference of 1.48 eV. These suggest that HOMO of phenols would prefer to interact with LUMO lobe on the carbonyl carbon of DMC via path A to provide the corresponding phenyl carbonates (Figure 4), and the interaction with LUMO+1 lobe on the methoxy carbon of DMC via path B to produce the corresponding O-methyl ethers will encounter higher energy barriers.
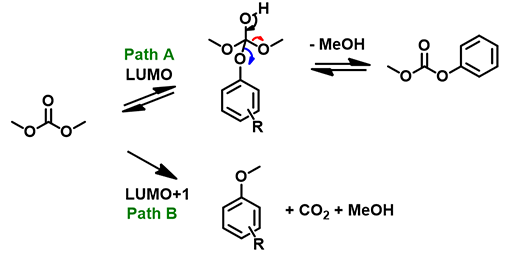
Figure 4: Two potential reaction paths between different phenols and DMC
p-Methoxyphenol, which is the least acidic phenol in the set, reacts with DMC to provide a 77:23 mixture of carboxymethylation and methylation products via path A and the higher energy path B, respectively, with the highest percentage of carboxymethylation reported in the study. In contrast, reaction with p-cyanophenol, which is the most acidic phenol in the paper, provides quantitatively and selectively, only the methylation product (Figures 4, 5). We reasoned that the tetrahedral dimethoxy(aryloxy)methanol intermediate formed with the most acidic p-cyanophenol reverts most readily back to starting phenol, which is then methylated irreversibly via path B, a higher energy process. The overall observed ratios of carboxymethylation vs methylation products are related to the reaction temperature, reaction time, pKa of the phenol itself (Figure 5), the reversibility and irreversibility of the competing reactions, and the amount of methanol remaining in the reacting mixture.
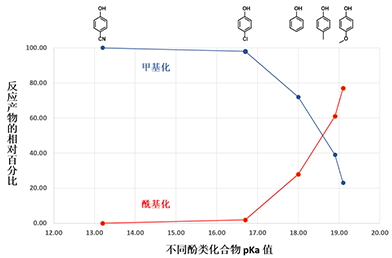
Figure 5: Plot of pKa of various phenols versus relative % of reaction products
Next, let’s look at the reactions of thiols and 1-octanol with DMC (Table 2).
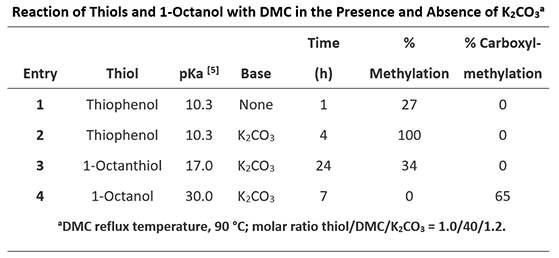
Table 2: Reaction of thiols and 1-octanol with DMC [2]
Thiols are more nucleophilic than alcohols, readily react with DMC, even in the absence of base. Thiol and thiophenol are significantly more acidic than the corresponding alcohol and phenol. With the tetrahedral intermediates formed by addition of thiophenol or thiol to the carbonyl group of DMC, thiol is a better leaving group than alcohol. Reverting of the tetrahedral intermediates back to dimethyl carbonate is more favorable than generating the thioesters, which are more reactive than DMC; as such, carboxymethylation of thiols was not observed (Figure 6). 1-Octanol interacts with DMC to reach an equilibrium mixture of DMC and transesterification products (methyl octyl and dioctyl carbonates). Since 1-octanol (pKa of 30.0) is not acidic enough to be deprotonated with potassium carbonate, no methylation product was observed.
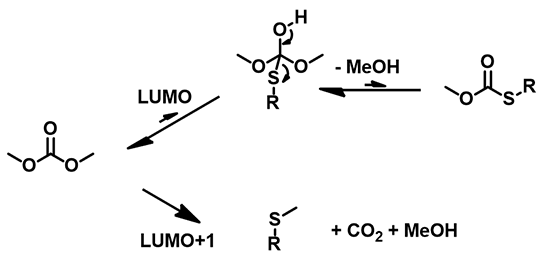
Figure 6: Potential reaction paths between thiols and DMC
Finally, let’s look at the reactions of aniline with DMC (Table 3).
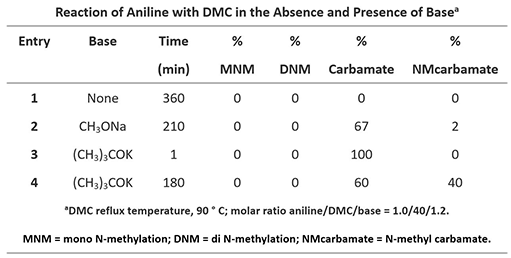
Table 3: Reaction of Aniline with DMC [2]
Reaction of aniline and DMC exhibited very different patterns as compared with those with phenols and thiols. Aniline does not react with DMC in the absence of base. When CH3ONa is used as the base, aniline reacts rapidly with the carbonyl carbon of DMC to form a relatively more stable carbamate and the reaction is irreversible (Figure 7, Path A-I). Methylation of aniline with DMC (Path B-I) will require using LUMO+1 of DMC with a higher energy barrier, not competitive with the irreversible acylation. The carbamate a might be further acylated to form intermediate b via Path A-II (LUMO), but due to its high reactivity towards methanol generated in the reaction, it’ll readily revert back to a and is not observed. Furthermore, since the carbamate NH is more acidic than that of NH2 on aniline, a will undergo deprotonation, then methylation to give c, via Path B-II (LUMO+1), an irreversible step. With (CH3)3COK, a stronger base, and extended reaction time, c continued to accumulate, until the reaction was stopped.
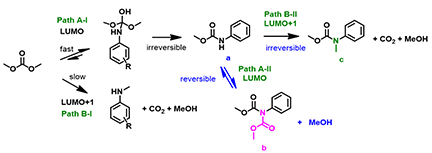
Figure 7: Potential Reaction Paths between Aniline and DMC
It is easier to understand the complex ambident reactivity pattern of dimethyl carbonate from the perspective of LUMO, pKa, reversibility and irreversibility of the reactions. Our insights enable us to predict how DMC will react under specific conditions. If you are interested in pKa estimation and LUMO analyses of electrophiles, please refer to Chapter 3 and Chapter 12 of our QM articles.
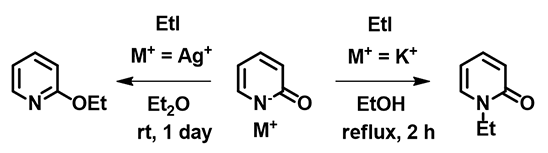
Figure 8: Alkylation of Pyridone
Let us now consider the results of the pyridone ethylation shown in Figure 8. In 2010, Mayr et al. pointed out alkylation results under silver salt conditions could not be accounted for with the SN1 carbonium ion mechanism [7]. Based on the calculated electron density map [8] below, what will you think a reasonable explanation could be? We also encourage you to read Mayr's excellent review article [3], say "Farewell to the HSAB concept", and analyze reaction of ambident reagents from thermodynamics and kinetics perspectives.
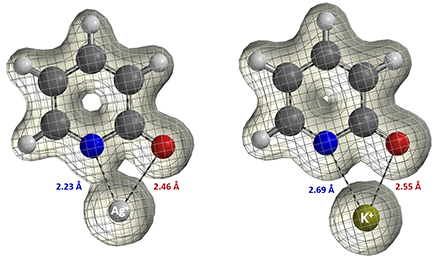
Figure 9: Calculated Electron Density maps of Pyridone Silver and Potassium Salts (Isovalue: Ag+ 0.046 e/au3, 92.0%; K+ 0.030 e/au3, 92.0%) [6, 8]
References:
[1] Pearson, R.G. J. Am. Chem. Soc. 1963, 3533.
[2] Tundo, P., Rossi, L., Loris, A. J. Org. Chem. 2005, 70, 2219.
[3] Mayr, H., Breugst, M., Ofial A. R. Angew. Chem. Int. Ed. Engl. 2011, 6470.
“Farewell to the HSAB Treatment of Ambident Reactivity: We have analyzed literature reports on the regio-selectivities of these and other ambident nucleophiles and electrophiles, and we have realized that the number of cases where the HSAB principle and the concept of charge- and orbital-controlled reactions give correct predictions approximate the number of cases where they fail. For that reason, we suggest abandoning these concepts as guides for predicting ambident reactivity."
[4] Drago, R.S. J. Chem. Educ. 1974, 51, 300 and references 6 – 9 cited in Mayr, H., Breugst, M., Ofial, A. R. Angew. Chem. Int. Ed. Engl. 2010, 6470.
[5] A: https://organicchemistrydata.org/hansreich/resources/pka/pka_data/pka-compilation-reich-bordwell.pdf
B: https://organicchemistrydata.org/hansreich/resources/pka/#kaalcohol
C: https://organicchemistrydata.org/hansreich/resources/pka/#kathiol
[6] https://www.wavefun.com/spartan-documentation
[7] Breugst, M., Mayr, H. J. Am. Chem. Soc. 2010, 15380.
[8] Johnson, E. R., Keinan, S., Mori-Sánchez, P., Contreras-García, J., Aron, J., Cohen, A. J., Yang, W. J. Am. Chem. Soc. 2010, 6498.
This article is written and edited by Shouliang Wang, Jian Wang, Qiuyue Wang, Zhong Zheng, Guqin Shi, Yongsheng Chen, and John S. Wai.