 Choose language
Choose language
< Return to main menu
Choose language



News & Events

Services & Solutions
News & Events
Career
Platform
Magical Power of Quantum Mechanics
Events
News
Magical Power of Quantum Mechanics
In previous chapters, we discussed the application of HOMO and LUMO for predicting regioselectivity of EAS/SNAr reactions of heterocyclic aromatic substrates, and calculated 13C NMR for structure assignments. Below is an example on how these QM calculations are integrated into our routine chemistry workflow [1].
For the synthesis of target molecule 4, we envisioned a C-2 selective bromination of starting material imidazo[1,2-a]pyrazine 1 to provide the bromo intermediate 2 (Figure 1), followed by a bromide-Grignard exchange and addition of acetaldehyde to afford intermediate compound 3, then a selective SNAr reaction of 3 with methyl amine at C-6. Next, we evaluated feasibility of the sequence with QM.
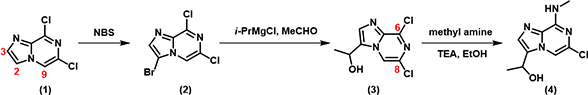
Figure 1: Designed synthetic route of target molecule 4
From Chapter 2 , we learned that regioselectivity of EAS halogenation[2,3] could be reliably predicted using calculated HOMO, and 13C NMR.
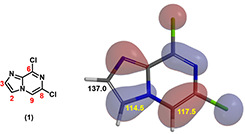
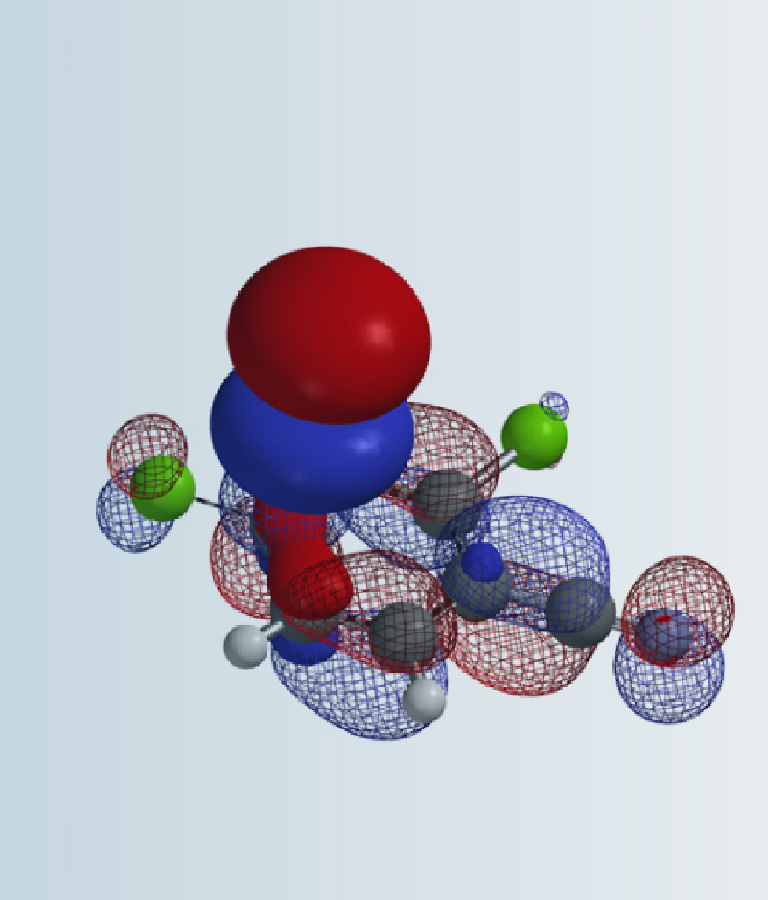
Figure 2: HOMO and calculated 13C Chemical shift of compound 1
HOMO of compound 1 shows no HOMO lobe at C-3 position, while a big one over C-2 position (Figure 1), suggesting a preference for C-2 regioselectivity of halogenation on the imidazole ring. Furthermore, calculated 13C chemical shift of C-2 (114.5 ppm) and C-3 (137.0 ppm) showed significant difference, with a lower chemical shift on C-2, indicating a higher electron density on the carbon and C-2 halogenation to dominate. Both HOMO and 13C NMR point to selective reaction at C-2 over C-3.
On the other hand, HOMO lobe on C-9 position of compound 1 is comparable in size to C-2 of the imidazole ring, and 13C NMR chemical shifts only differ by 3 ppm – will we run the risk of having competing halogenation on C-2 and C-9? In general, as imidazole is electron-rich while pyrazine is electron-deficient, electrophilic substitution reactions are more likely to proceed on the more electron-rich imidazole. Do we have additional QM calculation tools that confer us with a higher level of confidence on the desired regio-selectivity before we start chemistry in the lab?
In Chapter 17 , we introduced the use of relative energy of halonium ion as the third QM parameter for such prediction. For substrate 1, calculated relative energy of brominium ion (Equilibrium Geometry) for the three potential substitutable sites, C-2, C-3 and C-9, are tabulated in Figure 3. The relative energy of 2B is much lower than that of 2C and 2A, suggesting the bromination should occur only at C-2 position. And indeed, this is what we observed. Although intermediate 2 has multiple halogen atoms, we reasoned that the rate of Br-Mg exchange is much faster than that of Cl-Mg exchange, and the formation of intermediate 3 will be highly selective.
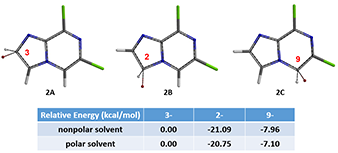
Figure 3: Equilibrium Geometry and Relative Energy of C-3, C-2, C-9 Brominium ions
The next question is on the selectivity of the SNAr reaction of intermediate 3. Which chloride will be preferentially displaced by the amine? In Chapter 1 , we learned that nucleophile will preferentially attack the carbon with larger LUMO lobe.
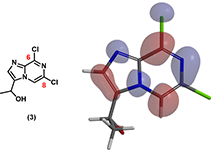
Figure 4: LUMO of intermediate 3
LUMO of intermediate 3 shows significant lobe over C-6 and close to none over C-8, suggesting that the nucleophile would react preferentially on C-6 position. This is what we observed experimentally.
In Chapter 11 “QM Assisted Structure Assignment with 13C NMR Calculation”, we learned to use Quantum Mechanics calculated versus experimental 13C NMR chemical shift for structure assignment. In this example, we applied such analysis to be assured that we indeed obtained target 4 (Figure 5), not any of the other potential isomers, 4A, 4B or 4C [4, 5]. The calculated 13C NMR data of structure 4 matched the experimental data the best versus the other isomeric structures, enabling us to assign the structure of compound 4 with high confidence.
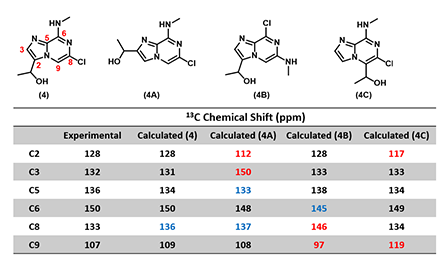
Figure 5: Experimental 13C NMR of the final product 4 and the calculated data of structural isomers (Spartan '18)
(Note: black: Δ ≤ 2 ppm, blue: 2 < Δ ≤ 10 ppm, red indicates: Δ > 10 ppm.)
In summary, we integrate multiple QM applications to evaluate feasibility of synthetic routes and compare calculated versus experimental 13C NMR for accurate structure assignment of the final targets. Quantum Mechanics give us Insight and Numbers, changing the ways we analyze chemistry retrospectively and prospectively. [6]
For bromination of compound 5 with NBS, let’s look at QM calculated HOMO, 13C NMR and relative energy of the three potential brominium ions (Equilibrium Geometry) on Figure 6. We could predict with high confidence which isomer will be the only product produced.
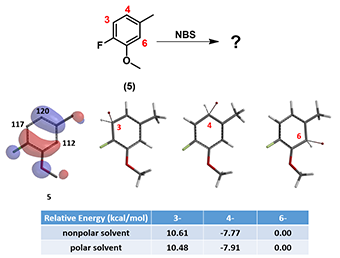
Figure 6: HOMO, 13C NMR of compound 5 and equilibrium geometry-relative energies of C-3, C-4, and C-6 brominium ions.
This article is written and edited by Chunrui Wu, Guqin Shi, Qiuyue Wang, Dong Pan, Yongsheng Chen, and John S. Wai.
References:
[1] J. H. Warren. A Guide to Molecular Mechanics and Quantum Chemical Calculations. Irvine, CA, USA: Wavefunction, Inc., 2003.
[2] F. A. Carey & R. J. Sundberg. Advanced Organic Chemistry Part A: Structure and Mechanisms. New York, NY, USA: Springer Science + Business Media, LLC., 2000. Pg. 551-557.
[3] M. Kruszyk, M. Jessing, J. L. Kristensen, M. Jorgensen, J. Org. Chem. 2016, 81, 5128.
[4] M. Lodewyk, M. Siebert, D. Tantillo, Chem. Rev. 2012, 112, 1839.
[5] W. Hehre, P. Klunzinger, B. Deppmeier, A. Driessen, N. Uchida, M. Hashimoto, E. Fukushi, Y. Takata, J. Nat. Prod. 2019, 82, 2299.
[6] F. Neese, M. Atanasov, G. Bistoni, D. Maganas, S. Ye, J. Am. Chem. Soc., 2019, 141, 2814.