 Choose language
Choose language
< Return to main menu
Choose language



News & Events

Services & Solutions
News & Events
Career
Platform
Magical Power of Quantum Mechanics
Events
News
Magical Power of Quantum Mechanics
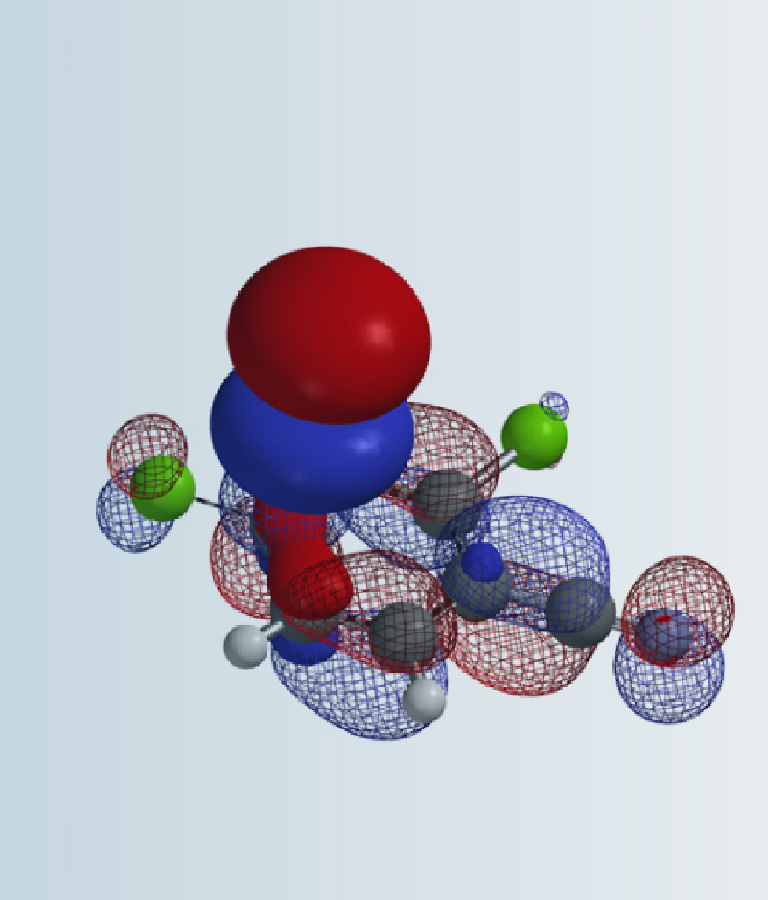
Chapter 2, left us with a unique substrate with HOMO lobes very close in size at C6 and C7 positions, and no significant difference in the 13C chemical shift. This is consistent with the electron donating characteristic of the amide and ether functionalities on the phenyl ring, and led us to expect a mixture of C6 and C7 brominating products. But experimentally only C7 bromide was obtained. How could we account for this?
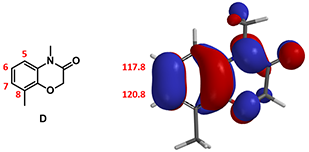
Figure 1: Schematic of HOMO & 13C NMR of compound D
From Chapter 9 we learned the use of calculated activation energy to compare two competing reaction paths. Let’s do this first.
To save computational time, we substituted NCS for NBS. The activation energies calculated for halogenation at C6 and C7 positions are 36.21 and 34.08 kcal/mol, respectively (Figure 2), suggesting that the reaction occurs more readily at the C7 position.
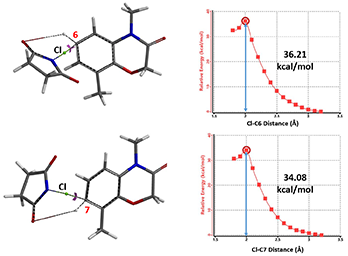
Figure 2: Calculation of Halogenation Activation Energy at C6 and C7
Based on Arrhenius equation (Figure 3), an activation energy difference of 2.13 kcal/mol translates to a ratio of 113:1, accounting for why only C7 brominated product is observed.

Figure 3: Calculating product ratio with Arrhenius Equation
In general, activation energy calculations require relative accurate model of the reaction and longer computation time [1, 2]. Do we have other more convenient, faster methods to predict accurately regioselectivity of such halogenation reactions?
In 2018, Jørgensen et al. reported in Chem. Sci. [3] the development of RegioSQM method for rapid prediction of the regioselectivity of electrophilic aromatic substitution reactions. RegioSQM works by protonating all aromatic C–H carbon atoms and identifying those with the lowest free energies in chloroform using the PM3 semiempirical method as the most nucleophilic center (Figure 4). When RegioSQM was used to predict regioselectivity for compound D, it also predicted a mixture of halogenation products.
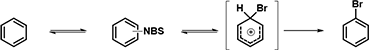
Figure 4: Classical Halogenation Reaction Mechanism
We reasoned that Jørgensen et al. method could be improved by calculating Equilibrium Geometry of the relevant halonium ions with a more accurate QM method (with DFT ωB97X-D 6-31G* [4]) and comparing their relative energies to predict selectivity of the halogenation.
Relative energies from Equilibrium Geometry calculation of the C5, C6, and C7 brominium ions revealed that the C7 one has the lowest energy, in both polar and nonpolar solvents, suggesting that the brominated product at position C7 would be the major product (Figure 5). Consistent with observation.
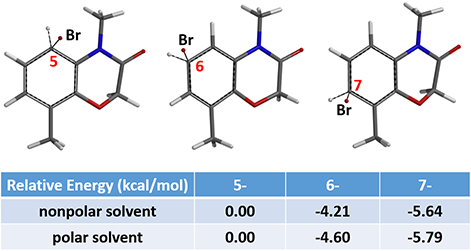
Figure 5: Equilibrium Geometry and relative energy of brominium ion at positions C5, C6, and C7
In summary, for electrophilic halogenation, we could use activation energy calculations to find transition states, and predict regioselectivity and product ratio. However, such calculations are time-consuming. A faster alternative, with comparable accuracy, is calculating for Equilibrium Geometry and comparing the relative energies of halonium ions for prediction.
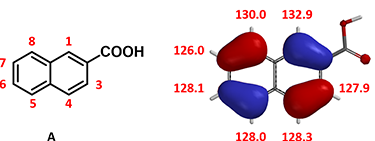
Figure 6: Calculated HOMO and 13C NMR of 2-naphthoic acid (A)
For predicting regioselectivity of the bromination of 2-naphthoic acid (A), we found that the HOMO lobe sizes of four of the seven sites are very similar, and the chemical shifts of the 13C NMR are not significantly different. Relative energies from Equilibrium Geometry calculation of the brominium structures are tabulated below. The major product is…..
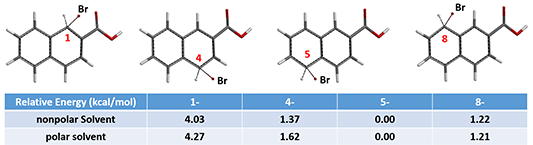
Figure 7: Equilibrium conformation and relative energy of brominated cations at C1, C4, C5, and C8.
This article is written and edited by Zhou Xu, Guqin Shi, Yongsheng Chen, and John S. Wai.
References:
[1] Hoffmann, R., Malrieu, J.P. Angew Chem Int Ed Engl., 2020, 59, 12590 (Simulation vs. Understanding, “To predict is not to explain.”)
[2] Structurally, halogenating reagent is an integral part of the transition state of halogenation. The successful use of relative energy difference between halonium ion to predict regioselectivity of halogenation does not necessary mean these simplified structures are the transition states.
[3] Jørgensen, M. et al, Chem. Sci., 2018, 9, 660. RegioSQM http://www.regiosqm.org
[4] Spartan '18 Tutorial and User's Guide (2019). Irvine, CA, USA: Wavefunction, Inc.