 Choose language
Choose language
< Return to main menu
Choose language



News & Events

Services & Solutions
News & Events
Career
Platform
Magical Power of Quantum Mechanics
Events
News
Magical Power of Quantum Mechanics
Bromination of 3-acetyl-5-hydroxyindole 1 provided selectively only the C6 brominated product 2, while Mannich reaction of it generated only the C4 substitution product 3 [1]. How to account for this dichotomy?
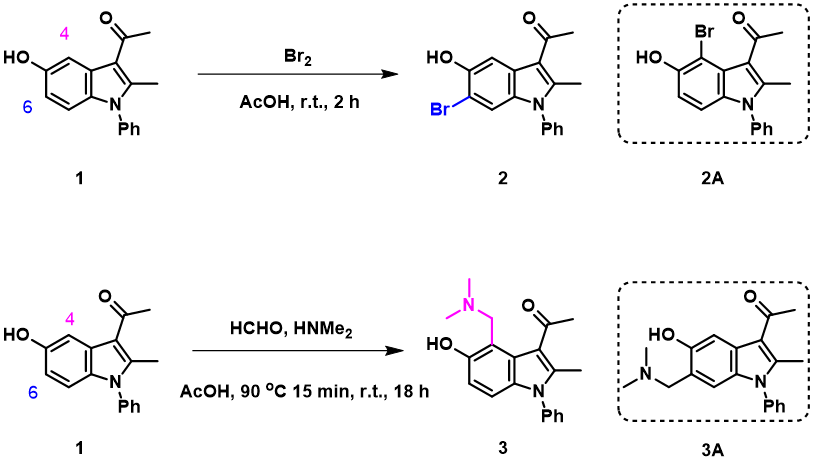
Figure 1. Bromination and Mannich reaction of 3-acetyl-5-hydroxyindole 1
In chapter 2 “Application of HOMO Analyses in Electrophilic Reaction“, we used the comparative difference in size of HOMO lobes and calculated 13C NMR for predicting regioselectivity of halogenation. As shown in Figure 2, energy gap between HOMO (-7.46 eV) and HOMO-1 (-7.57 eV) of substrate 1 is small, and the HOMO lobe at C4 is similar in size as the HOMO-1 lobe at C6, suggesting the reaction will provide a mixture of C4 and C6 products. Calculated 13C NMR chemical shift value is 104 ppm for C4 vs 111 pm for C6, suggesting that C4 bromide will be the major product. Obviously, the above QM parameters derived from equilibrium geometry of indole 1 fail to account for the experimental result.
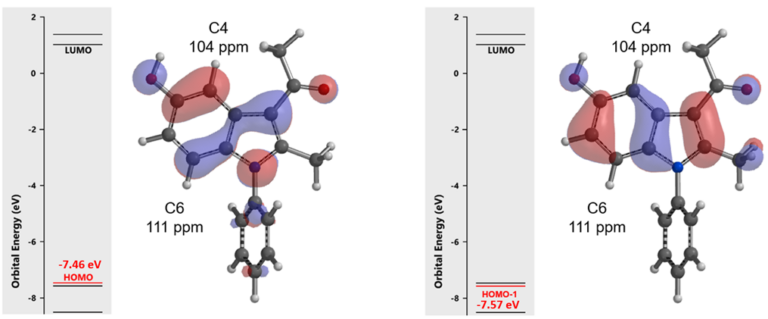
Figure 2. HOMO (left), HOMO-1 (right), calculated C4 & C6 13C NMR shifts of indole 1
In Chapter 17 “Predicting Regioselectivity of Electrophilic Halogenation Reactions“, we discussed the use of QM calculated Equilibrium Geometry of Wheland intermediates’ relative energy difference for predicting regioselectivity. The calculated relative energies of the C4, C4a, and C6 Wheland intermediates are significantly lower than the C7 one, with the C4a one slightly lower than the C6, suggesting the bromination will provide a mixture of C4 and C6 substitution products, with C4 bromide being the major one. This well-established model for electrophilic aromatic reaction fail to account for the experimental result as well.
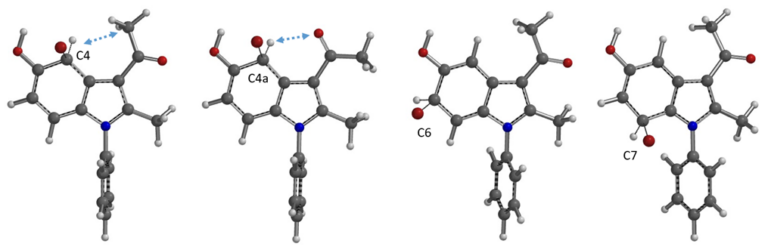
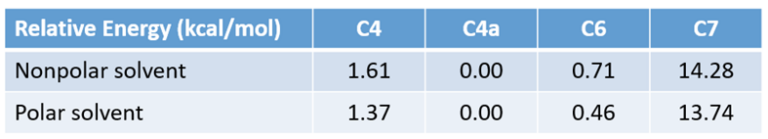
Figure 3. Equilibrium geometry and relative energy of bromo Wheland intermediates C4, C4a, C6, and C7
Why all these basic methods, differences in relative size of HOMO lobes, in 13C NMR shift, difference in relative energy of the hypothetical Wheland intermediates, fail to predict regioselectivity of bromination of indole 1? Shown in Figure 4 is a general mechanism for electrophilic aromatic substitution. This involves interaction of positively charged electrophile (E+) with the aromatic substrate to form the positively charged Wheland intermediate [2], followed by loss of proton (H+) or rearomatization to provide the bromination product. Formation of the Wheland intermediate is assumed to be the rate-determining step; the subsequent rearomatization is presumed to be a much faster process. We reasoned that with indole 1 steric hindrance may have slowed down the rearomatization step, rendering it the rate limiting step of the reaction. The established prediction methods are not applicable for indole 1. The question becomes, how to establish a more relevant model for this substrate?
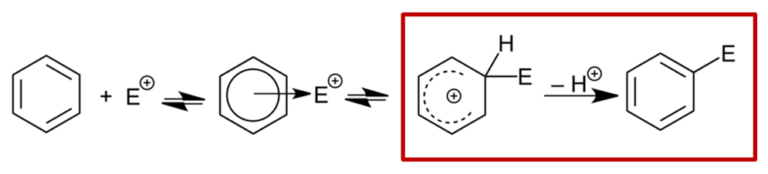
Figure 4. General mechanism for electrophilic aromatic substitution[2]
Since bromine was used as the brominating reagent in the reaction, Br2 will be the most appropriate for use to model its reaction with indole 1. Shown in Figure 5 is the calculated energy plot for C4 bromination. From Br2 and indole 1 (Reactants) to transition state 1 (TS1) towards formation of the transient Wheland Complex like structure (WC) requires an activation energy of 4.92 kcal/mol. The distal bromine then interacts with both the C-5 hydroxy group and the reaction center’s sp3 CHBr hydrogen to form a unique hydrogen bond stabilized Bromide-Wheland Complex (Bromide-WC), which is 4.05 kcal/mol lower in energy than the Reactants. The bromide in the 6-membered hydrogen bond ring of the Bromide-WC could deprotonate the sp3 CHBr hydrogen,[3] form HBr, and rearomatize to provide C4 bromination product. However, in transition state 2 (TS2), the sp3 CHBr bromide faces steric hindrance from the 3-acetyl group, as indicated with the calculated energy barrier of 11.59 kcal/mol, higher than the 8.92 kcal/mol needed to reverse back to the Reactants. This is likely to be the case as no C4 bromination was observed.
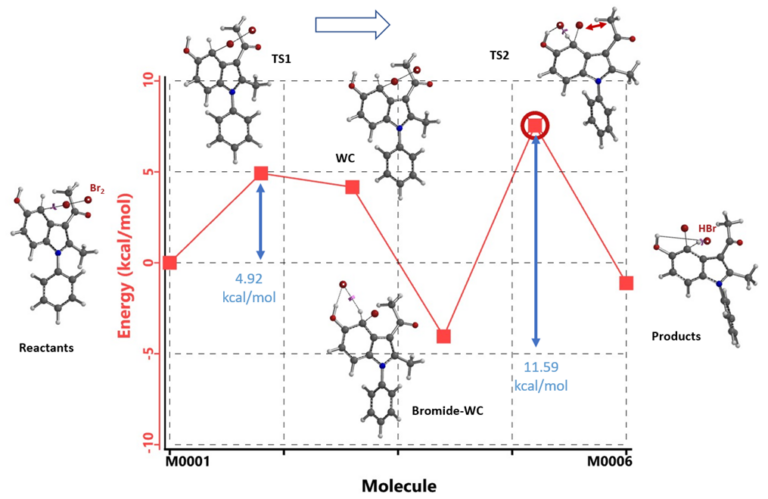
Figure 5. Calculated energy profile of indole 1 C4 bromination
Shown in Figure 6 is the calculated energy profile for C6-bromination. Br2 and indole 1 (Reactants) to TS1 towards formation of the transient WC requires an activation energy of 6.21 kcal/mol, significantly higher than 4.92 kcal/mol calculated for C4 bromination. The distal bromine then interacts with both the C-5 hydroxy group and the reaction center’s sp3 CHBr hydrogen to form a hydrogen bond stabilized Bromide-WC, which is 2.07 kcal/mol lower in energy than the Reactants. The bromide in the 6-membered hydrogen bond ring of the Bromide-WC deprotonates the sp3 CHBr hydrogen, forms HBr, and rearomatizes to provide the C6 bromination product. With no significant steric barrier in the process, this results in a significantly lower calculated energy barrier of 6.71 kcal/mol. It is the difference in energy barrier in this elimination step, 11.59 at C4 vs 6.71 at C6, a difference of 4.88 kcal/mol, that differentiates the two competing paths, leads to the highly selective C6 bromination observed.
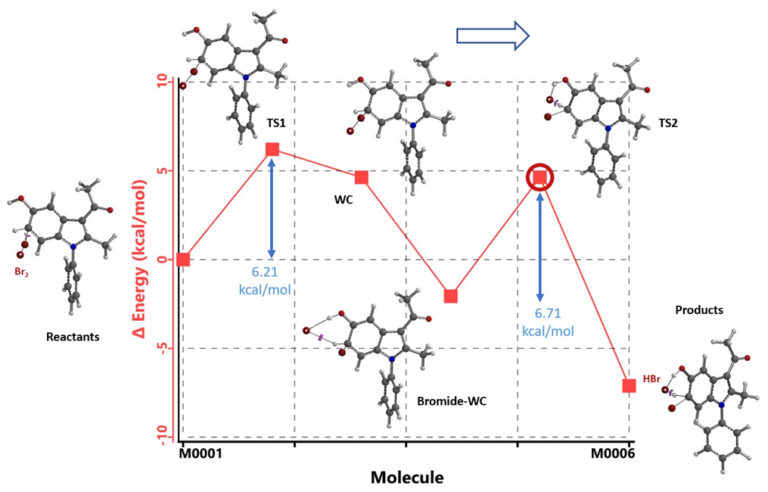
Figure 6. Calculated energy profile of indole 1 C6 bromination
In summary, calculations and comparison of the energy profiles of the potential pathways with full structure of the bromination reagent provide better insight on possible mechanism of the reaction and correlation with experimental observation. Reaction of indole 1 with N-bromo-succinamide (NBS) was reported to provide C6 selectively as well. Similar energy profiles could be generated by replacing the distal Br with succinimide in the calculations. For bromination in which the addition is the RLS, the positive charged, hypothetical, Wheland Complexes will be decent approximations; their relative energy usually correlates with experimental observations. While for bromination in which the elimination is the RLS, as shown in Figures 5 and 6, comparing the relative energy of the C4 vs C6 bromination products, 5.66 vs 0.00 kcal/mol respectively, may also be adequate for prospective regioselectivity analysis.
Mannich reaction of indole 1, in contrast to the highly selective bromination at C6 discussed above, provides only the C4 substitution product 3[1]. Both are electrophilic aromatic substitutions, shall regioselectivity of one be predictive of the other[2]?
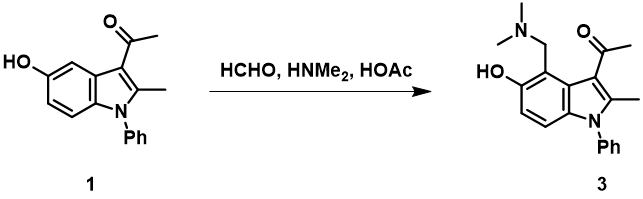
Figure 7. Mannich reaction of indole 1
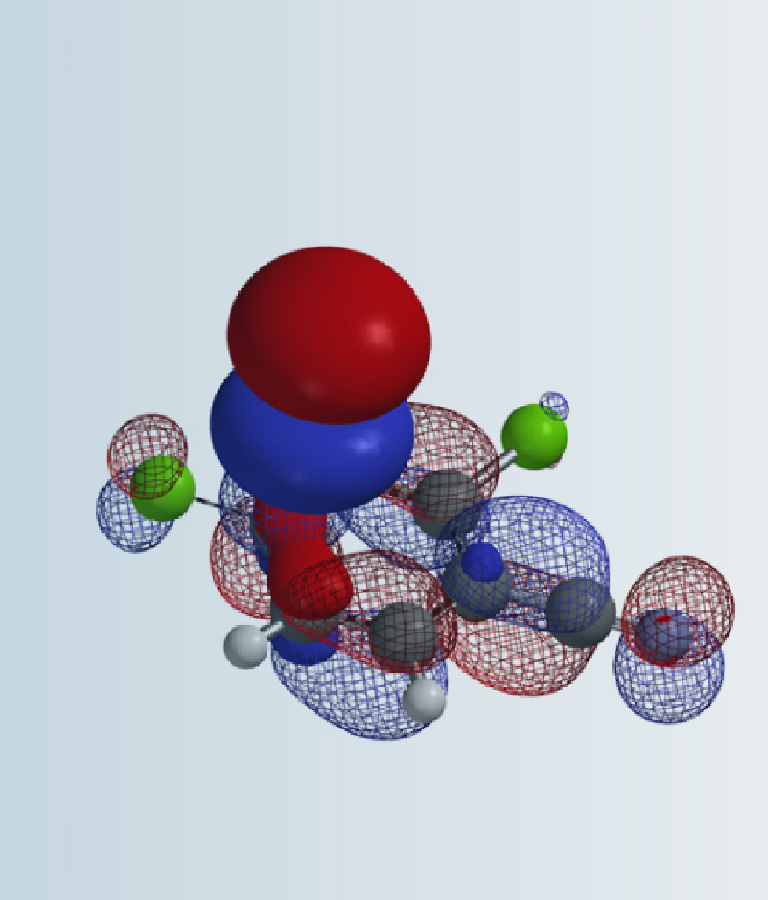
Formaldehyde and dimethyl amine react to form an iminium intermediate [H2C=N(CH3)2]+, a highly reactive species with LUMO energy of -5.24 eV. LUMO lobe on the iminium carbon is significantly larger than that of the nitrogen, accounting for selective nucleophilic attack on the iminium carbon (Figure 8, left). QM calculation reveals an accurate description of electron distribution of [H2C=N(CH3)2]+ (Figure 8, center), i.e. methyl H on iminium ion is positively charged, while the iminium nitrogen is negatively charged, instead of carrying a formal positive one as indicated in most organic chemistry textbooks[4]. Natural charge on nitrogen changes from -0.698 in dimethylamine to -0.275 in [H2C=N(CH3)2]+; the N becomes less negatively charged, not positively charged. Similar to tetramethylammonium ion described in QM chapter 23 (Figure 8, right)[5].
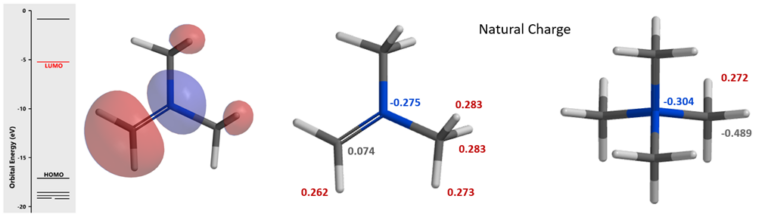
Figure 8. LUMO (left) and natural charge of [H2C=N(CH3)2]+ (center); natural charge of tetramethylammonium ion
Shown in Figure 9 (left) is transition state structure calculated for the Mannich reaction proceeding at C4 position, revealing a stabilizing Non-Covalent Interaction (NCI) between the C=O oxygen on 3-acetyl group of indole 1 and the methyl H on [H2C=N(CH3)2]+. On the other hand, this is not observed with corresponding transition state with C6 attack (Figure 9, right), leading to an energy difference of 1.74 kcal/mol between the two of them, favoring C4 attack, accounting for the selectivity observed.
Figure 9. Calculated transition state structures: C4 attack shows 3-acetyl group C=O non-covalent interaction with [H2C=N(CH3)2]+ methyl hydrogen (left with electron density contour); C4 attack does not have such NCI stabilization
Differences in regioselectivity between electrophilic aromatic substitutions are not common. For the indole substrate 1, bromination at C4 faces steric interaction with the 3-acetyl group, while the Mannich reaction at C4 encounters electronic stabilization effect between the acetyl group and incoming iminium ion, accounting for the divergence. QM analyses require us to examine carefully structures of the substrates and electrophiles for potential neighboring group interactions and choose relevant calculation models that are consistent with experimental conditions. This also involves critical examination of the basic reaction mechanisms and recognizing Wheland Complex could be just another convenient conceptual approximation we learned.
Bromination of 8-methoxyquinoline with NBS provides only the C-5 product and could be accounted for with QM calculated HOMO, 13C chemical shifts, and relative energy of C-5 vs C-7 equilibrium geometry of the brominium ions. However, NBS bromination of analogous 8-hydroxyquinoline, with almost the same calculated QM parameters, generates a mixture of C-5 and C-7 bromides, with C-7 substituted one being the major product.
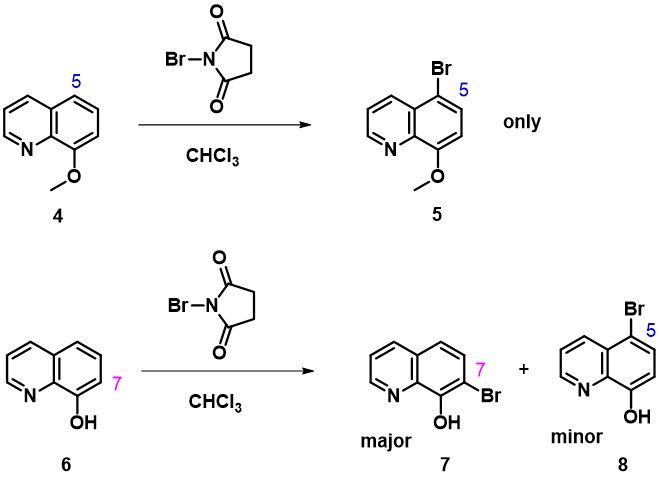
Figure 10. NBS Bromination of 8-methoxyquinoline and 8-hydroxyquinoline
We looked for potential hydrogen bonding interaction between the OH group and the brominating reagent to account for the divergence. Extensive QM analyses led to our current hypothesis that relative overall rate of C-5 vs C-7 bromination is controlled by the rate of deprotonation of the C-5 and C-7 Wheland intermediates which are in reversible equilibrium as discussed above[2].
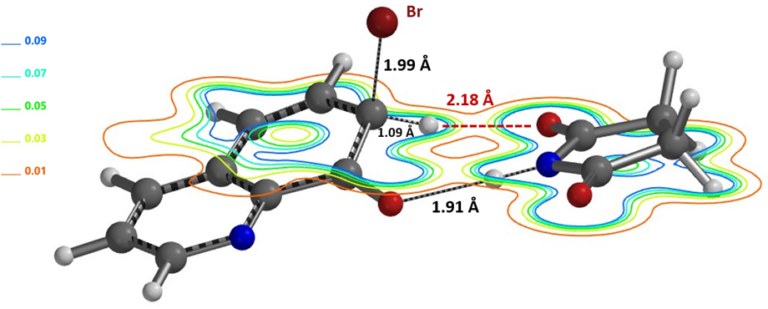
Figure 11. Equilibrium Geometry of 8-hydroxyquinoline-NBS complex with Electron Density Contour on the HO=CN plane.
Obvious questions arise. What is the role of succinimide anion (Figure 11)? Shall we expect a difference in C-7 vs C-5 product ratio under different reaction conditions, e.g., NBS vs bromine with different counter anion, under different reaction concentration, etc.
Jun Liu, Jianhui Qiao, Qiuyue Wang, Liting Dong, Yongsheng Chen, John S. Wai
References:
[1] G.S. Gadaginamath, A. G. Kamat, B. G. Pujar, Revue Roumaine de Chimie, 1995, 40, 265. Both Br2 and NBS brominating reagents reacts with indole 1 to provide selectively the C-6 bromide.
[2] https://en.wikipedia.org/wiki/Electrophilic_aromatic_substitution.
[3] W.J. Hehre, A.J. Shusterman, J.E. Nelson (1998) The Molecular Modeling Workbook for Organic Chemistry. Irvine, CA, USA: Wavefunction, Inc.; pp 86-7. Similarities between deprotonation and Sn2 reaction.
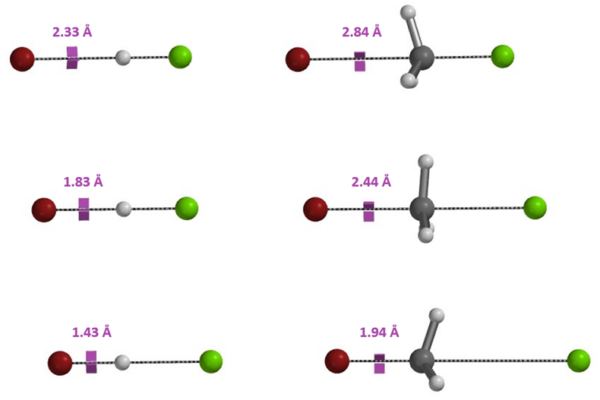
Figure 12. Br– approaches the hydrogen in HCl (left) and the carbon in CH3Cl (right) from the “backside”, displacing Cl–. Charge is transferred from Br– to Cl–
[4] E.V. Anslyn, D.A. Dougherty (2006) Modern Physical Organic Chemistry. New York, NY, USA: University Science Books; page 7 correctly points out the misrepresentation in most organic chemistry textbooks.
[5] QM Chapter 23 “A QM Study of the para Regioselectivity of TBABr3 Bromination” https://wuxibiology.com/a-qm-study-of-the-para-regioselectivity-of-tbabr3-bromination/
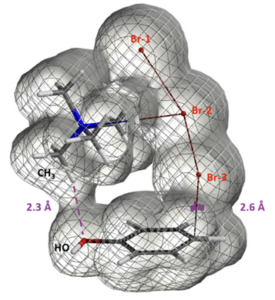
Figure 13. Electron density map (Isovalue 0.00744 e/au3, 98.0%) of TMABr3 – PhOH reaction complex with C – Br distance at 2.6 Å