选择语言
选择语言



新闻和活动

关于我们
服务和解决方案
新闻和活动
职业发展
服务平台
QM有机化学课堂
市场活动
新闻
QM有机化学课堂
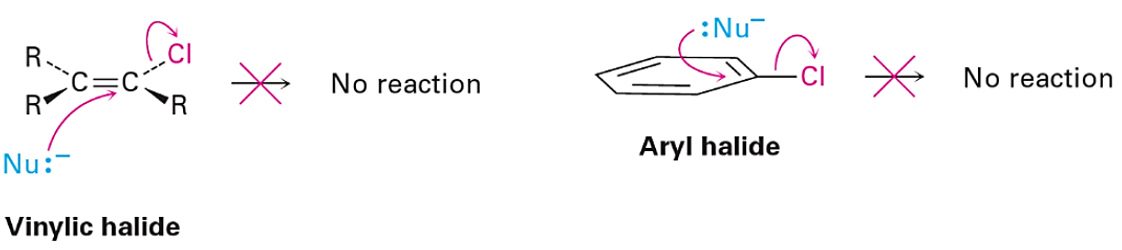
氯乙烷和氯乙烯都是重要的有机合成中间体,在有机合成中有着广泛的应用。图1所示为二者的LUMO和LUMO+1图,我们注意到氯乙烷的LUMO和氯乙烯的LUMO+1很相似,暗示它们应该可以发生一些相似的转化。事实也确实如此,例如二者均可以与镁反应制备相应的格氏试剂,强碱性条件下都可以消除一分子氯化氢生成乙烯或乙炔。氯乙烷分子中与氯原子相连的碳原子上有较明显的LUMO lobe分布,这解释了氯乙烷易于发生SN2亲核取代反应的原因。我们自然而然的想到了一个问题,氯乙烯是否也可以发生类似的平面内SN2反应呢?
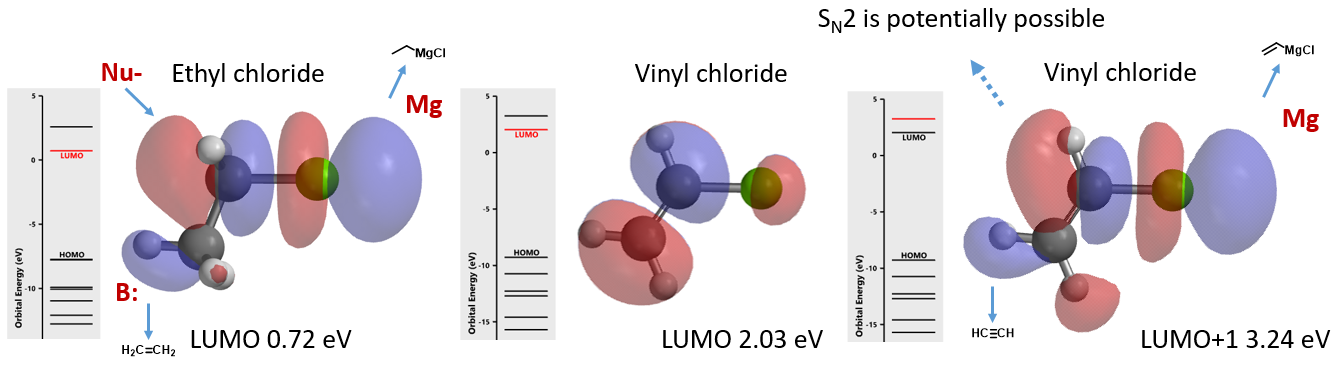
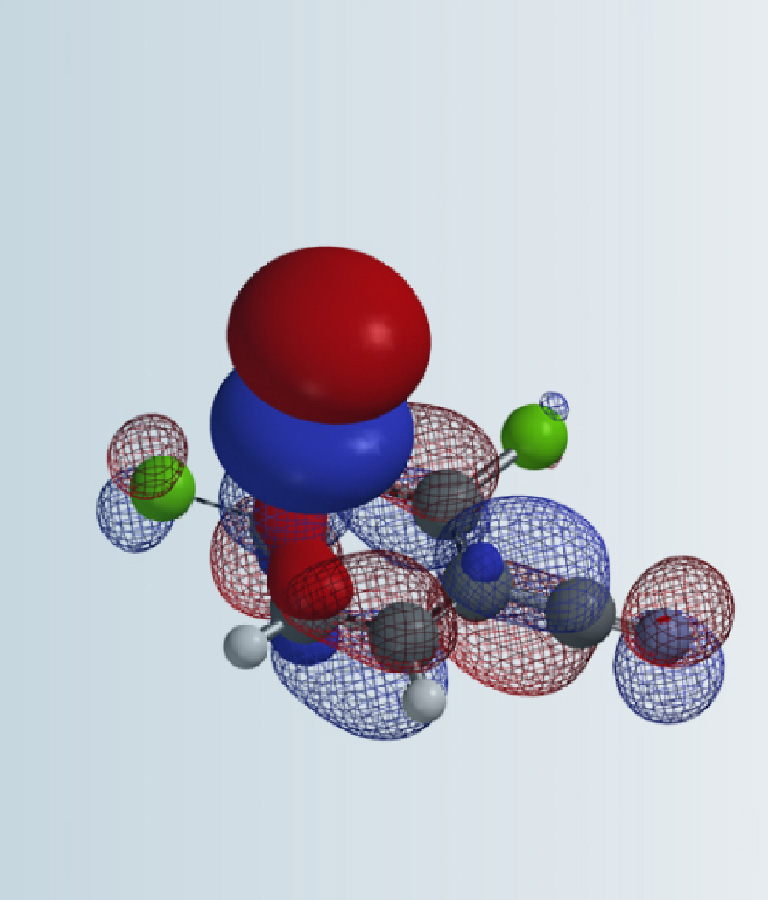
图1. 氯乙烷的LUMO及氯乙烯的LUMO、LUMO+1图
1994年Pross和Radom等人通过计算预测未活化的氯乙烯在气相和溶液中都存在发生平面内构型翻转的SN2亲核取代反应的可能性[1]。2004年,Ando和Narasaka等人利用密度泛函理论(DFT)计算首次预测化合物E-1可以发生分子内的平面内SN2反应,并通过实验进行了验证(图2)[2]。反应对底物的构型有严格的要求,E式底物在DMF溶剂中用NaH处理,反应在室温下就可以发生,以极高的收率得到苯并呋喃产物。而对于Z式底物,即使加热至110 ºC,仍然不会发生关环反应,几乎定量的回收原料。
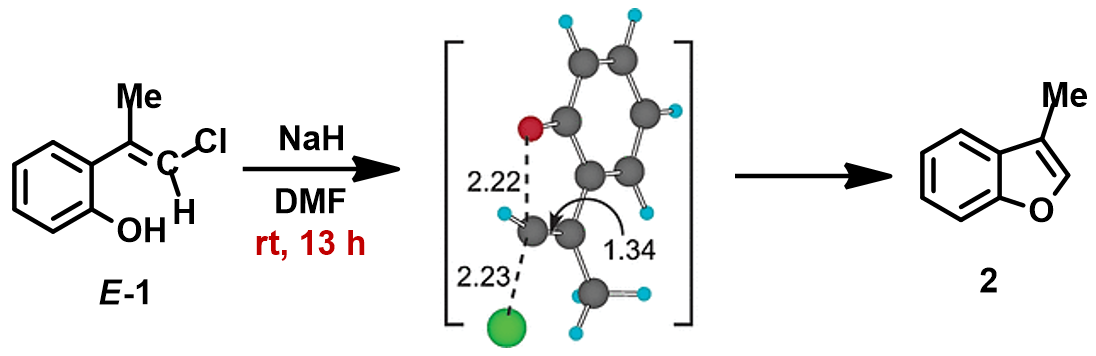
图2. E-1分子内面内SN2反应
作者使用B3LYP/6-31+G* (Onsager continuum, DMF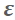 =37)方法计算得到了反应的过渡态结构(图2),但没有提供相关的坐标数据。我们重复了该过渡态计算,得到碳氧原子距离为2.158 Å和虚频为i556 cm-1的过渡态结构(图3),与文献报道值相近。
=37)方法计算得到了反应的过渡态结构(图2),但没有提供相关的坐标数据。我们重复了该过渡态计算,得到碳氧原子距离为2.158 Å和虚频为i556 cm-1的过渡态结构(图3),与文献报道值相近。
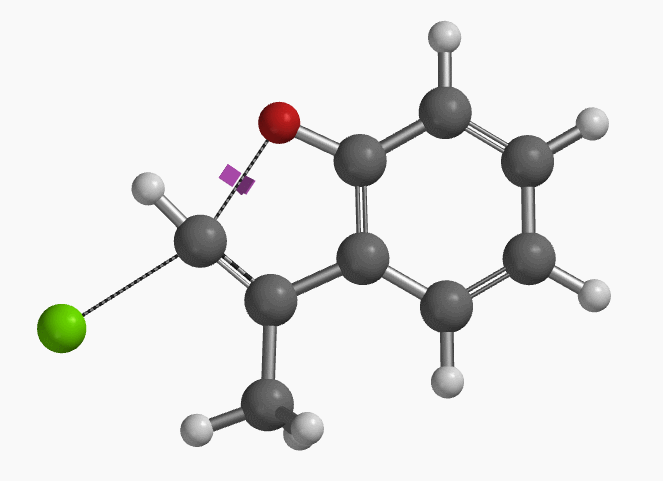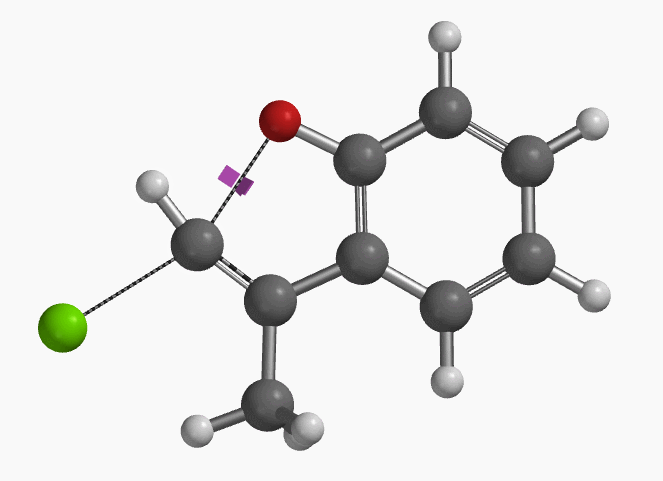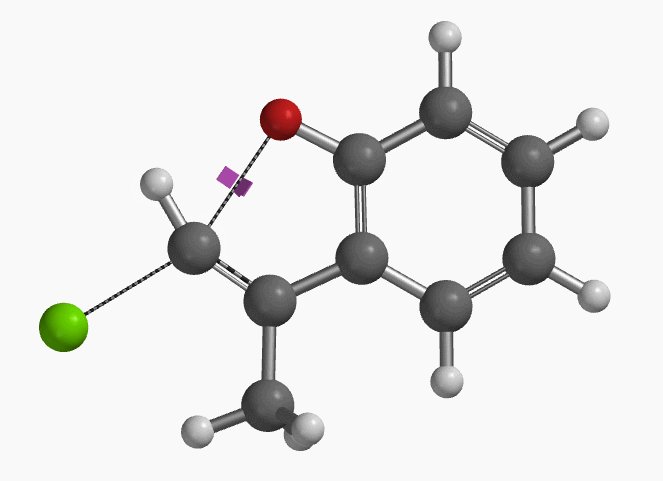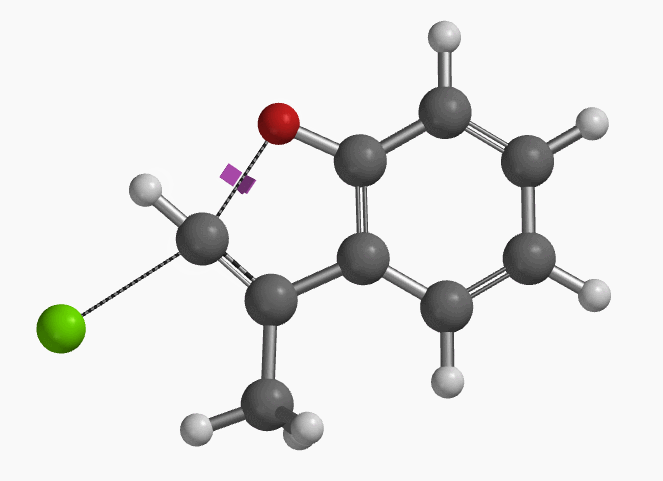
图3. E-1分子内面内SN2亲核取代反应的过渡态虚频i556 cm-1
我们通过内禀反应坐标(IRC) [3]计算得到了该反应历程的能量变化曲线(图4)。大家可以清楚的看到随着氧负离子的接近,乙烯基氢原子逐渐从与氧原子接近的内侧移动到外侧,氯离子逐渐离去。最终,碳氯键断裂,新的碳氧键形成,sp2中心碳原子发生了类似sp3碳原子在SN2反应中的伞状翻转,这一动图很好的展示了sp2碳原子参与的平面内构型翻转的SN2反应历程,这也很好的解释了为什么E式底物可以发生反应,而Z式底物却不能。E式底物中,氧原子与氯原子位于双键两侧,故而可以发生类似于经典SN2反应中亲核试剂(氧原子)从离去基团(氯原子)背面的进攻。而Z式底物中氧原子和氯原子位于双键同侧,所以反应难以进行。
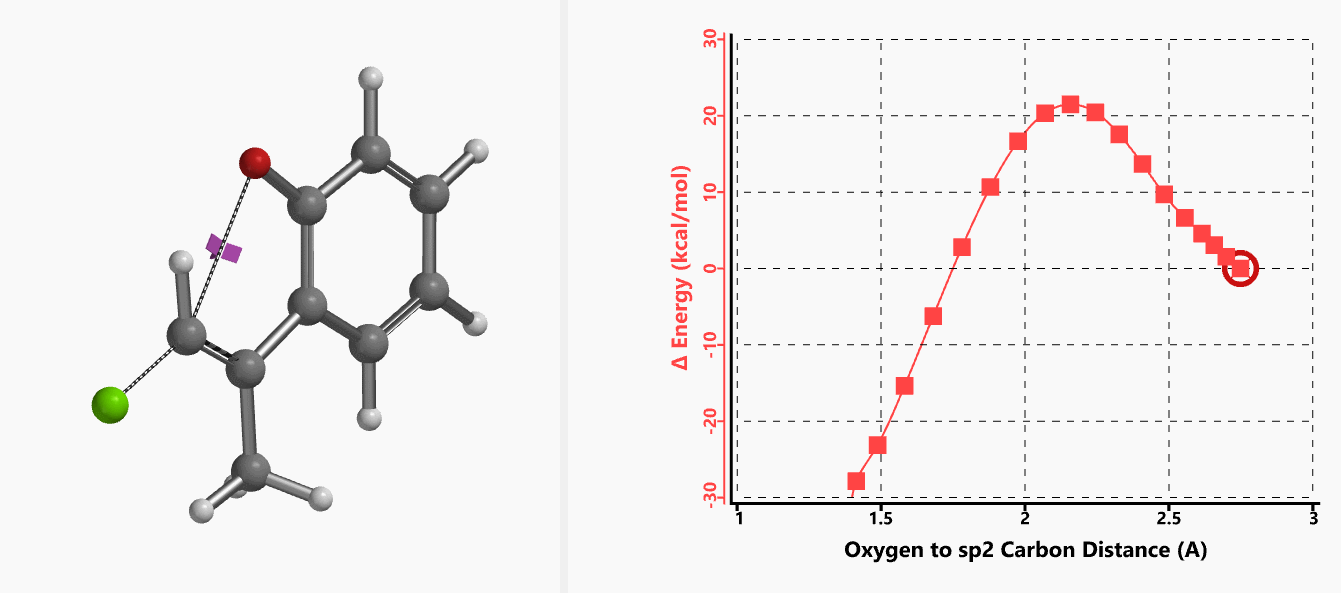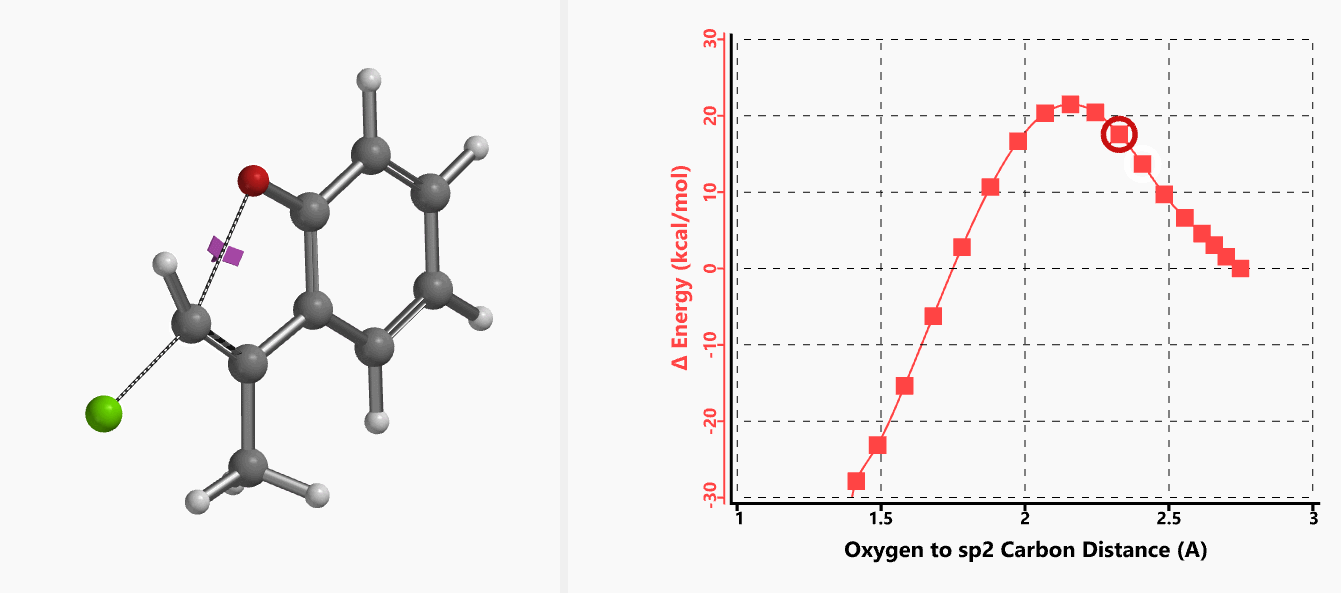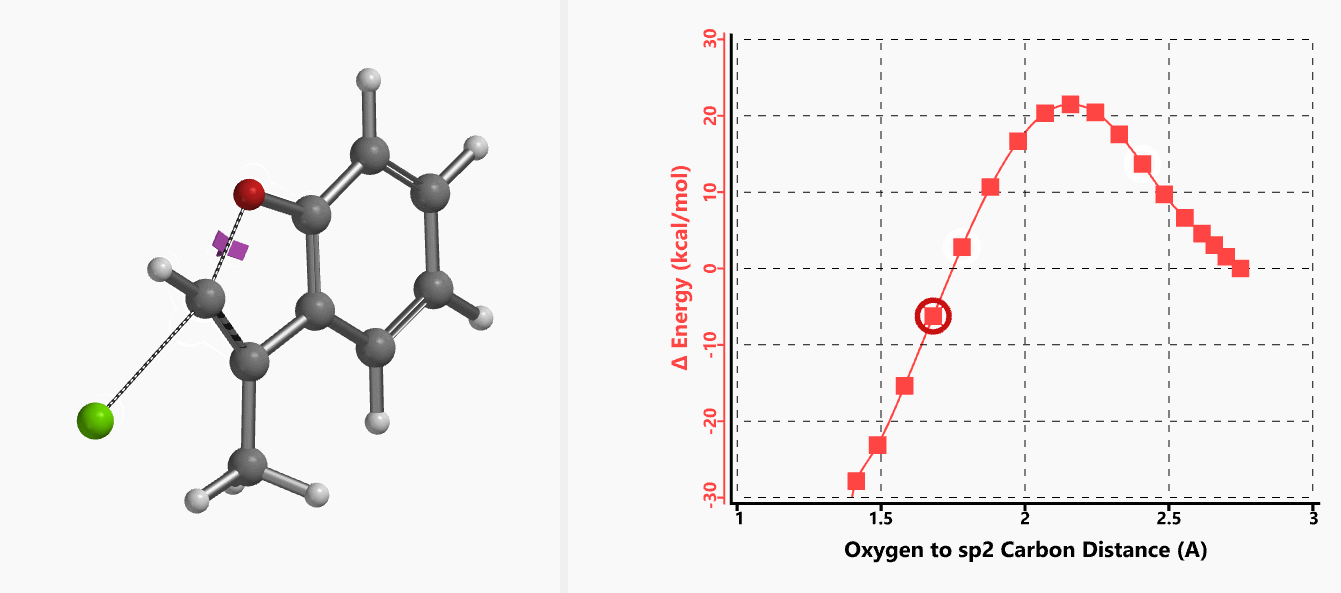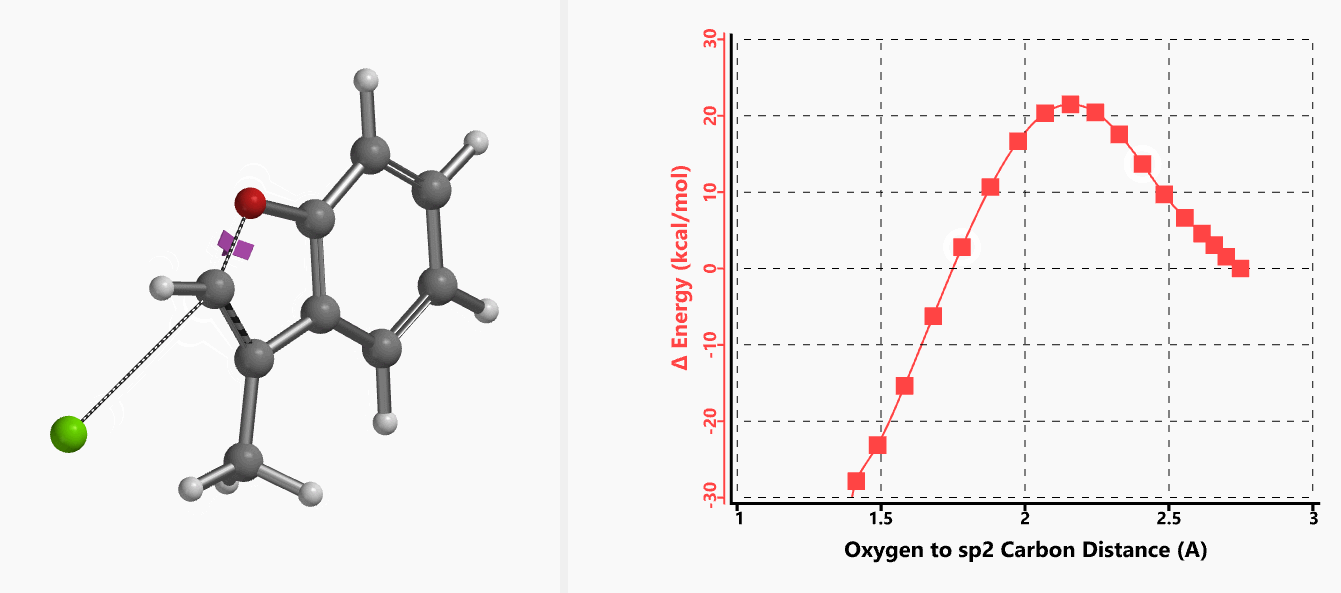
图4. E-1分子内面内SN2反应的能量变化曲线
图5所示为该反应能量变化曲线的等值线图,大家可以清晰的看到随着反应向过渡态的进行,氧负离子和乙烯基氢原子之间的电子密度逐渐增加。这一变化与氧原子和乙烯基氢原子之间的氢键距离相关,这种氢键作用使它们相互接近。当两原子间距离大约2.4 Å时,氢原子开始向另一侧翻转。氧原子和氢原子之间的电子云密度随之降低,而氧原子和sp2碳原子之间的电子云密度逐渐升高。
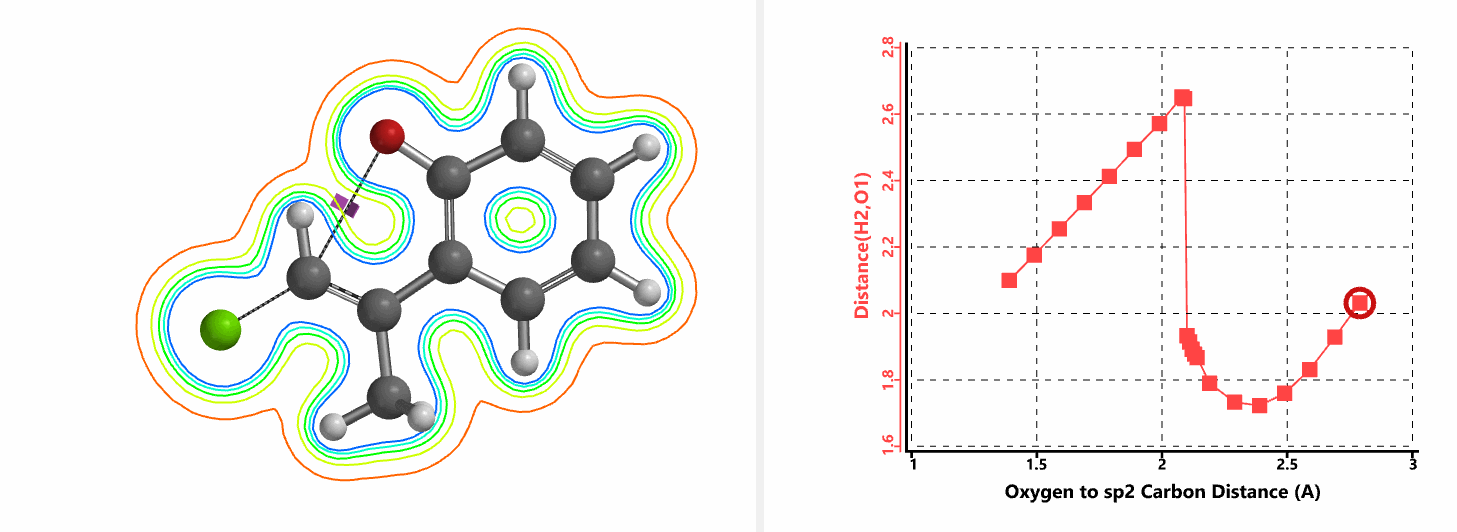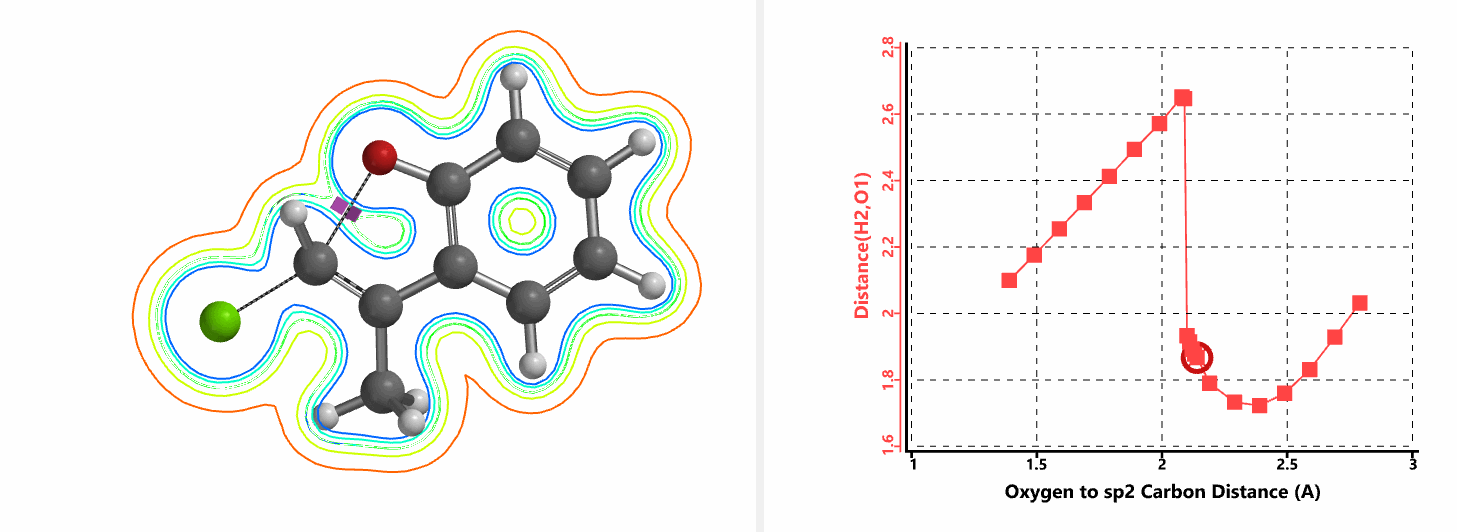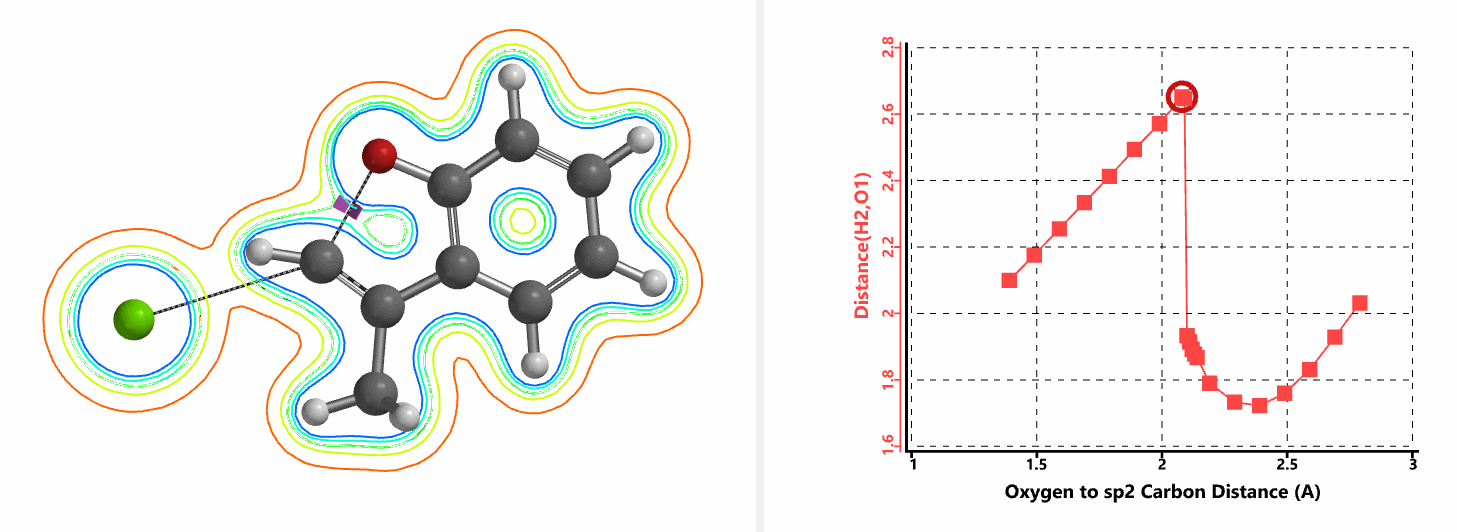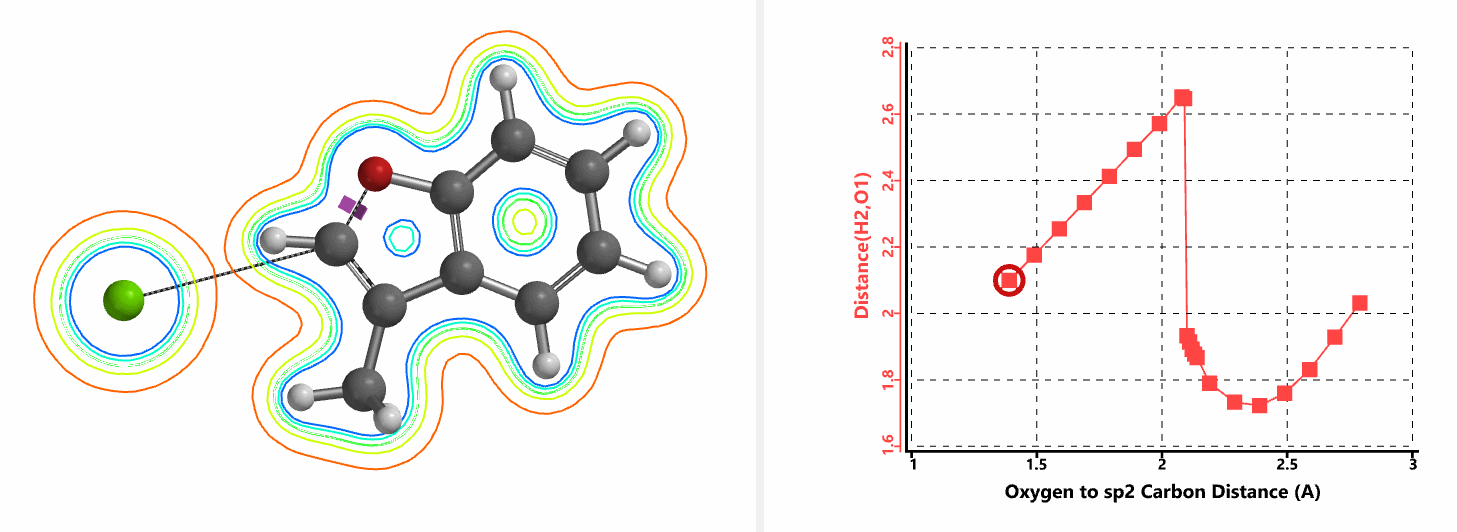
图5. E-1分子内面内SN2反应能量变化曲线的等值线图
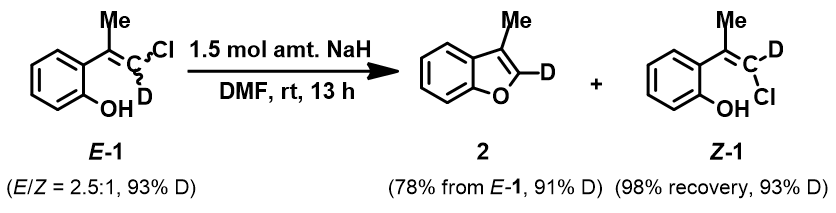
图6. 氘代E式和Z式混合物的分子内面内SN2亲核取代反应
当使用氘代氯烯烃的E式和Z式混合物作为底物时,相同条件下,反应得到氘保留的关环产物并几乎定量的回收到Z式氘代原料(图6)[4]。这一结果排除了反应是经过烯丙基异构化和卡宾插入机制发生的可能性(图7)。

图7. 烯丙基异构化和卡宾插入机制
此后,陆续出现了更多类似sp2平面内SN2亲核取代反应的文献报道,并且也都被计算支持[4]。
我们总结了一下,可以发生此类转化的底物类型(图8):
1. 亲核试剂中心原子可以为氧、氮、碳或硫原子[4]
2. sp2反应中心是碳原子。
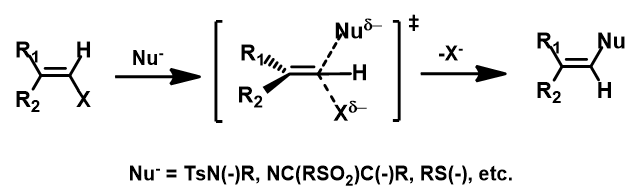
图8. sp2 SN2反应底物类型
前面讨论的都是发生在sp2碳原子上的SN2反应,接下来,我们一起看一个sp2氮原子的SN2亲核取代反应实例。2020年,Chen等人报道了一种高效的基于分子内氮氮键形成的2-芳基-1,2,3-三氮唑的合成方法[5]。作者使用碘甲烷选择性活化双腙化合物中的二甲胺基腙基团,然后用碱处理顺利地形成了多种2-芳基-1,2,3-三氮唑,产率从良好到极好。作者认为反应可能是通过SN2亲核取代机制进行的,其中三甲基胺作为良好的离去基团促进了分子内氮氮键的形成。如图9所示,碘甲烷活化的双腙底物3去质子化生成中间体4a,4a很容易互变异构为4b,随后通过分子内SN2取代反应生成三氮唑5。
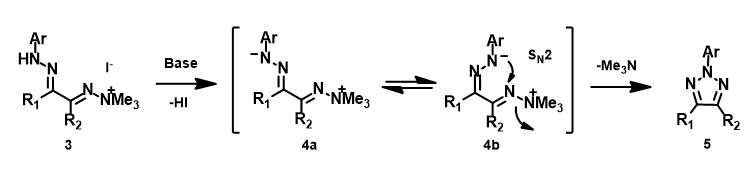
图9. 通过分子内 SN2 反应合成 2-取代-1,2,3-三氮唑
我们通过过渡态(iFreq i458 cm-1,图10)和内禀反应坐标(IRC)计算验证了作者提出的反应机制(图11)。如图11所示,反应中心原子为sp2杂化的氮原子,伴随着三甲基胺的逐渐离去,形成新键的两个氮原子慢慢接近并最终成键,反应历程为平面内构型翻转的SN2亲核取代反应。这一方法原料易得、反应条件温和且区域选择性高,是对sp2的SN2亲核取代反应的绝妙应用。如果通过Chan-Lam偶联反应制备这种2-芳基三氮唑则会得到混合物且反应收率低[6]。
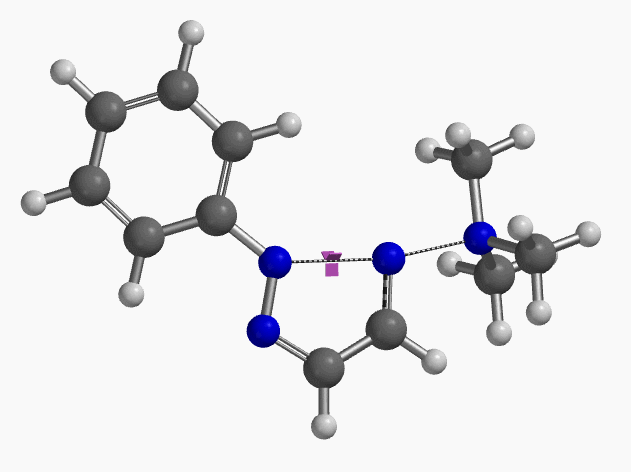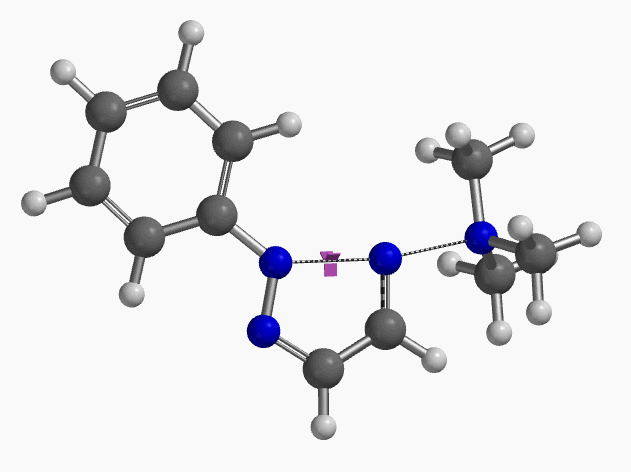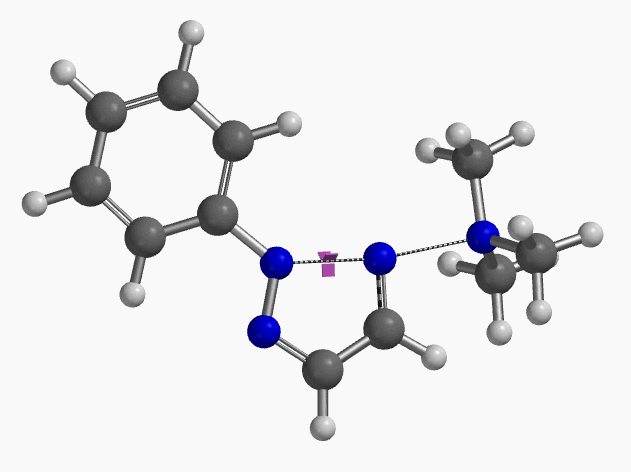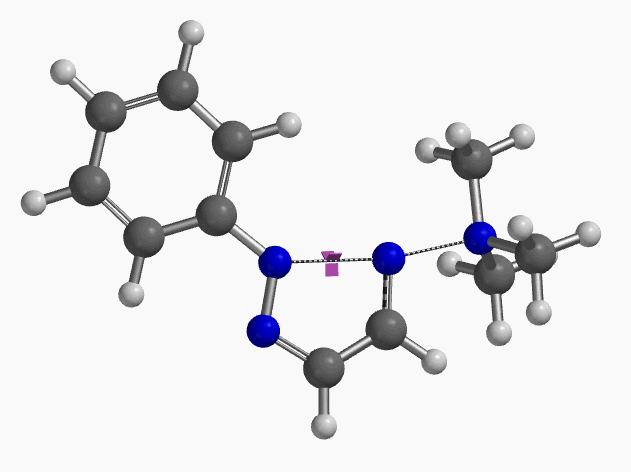
图10. 2-取代-1,2,3-三氮唑合成反应过渡态虚频i458 cm-1
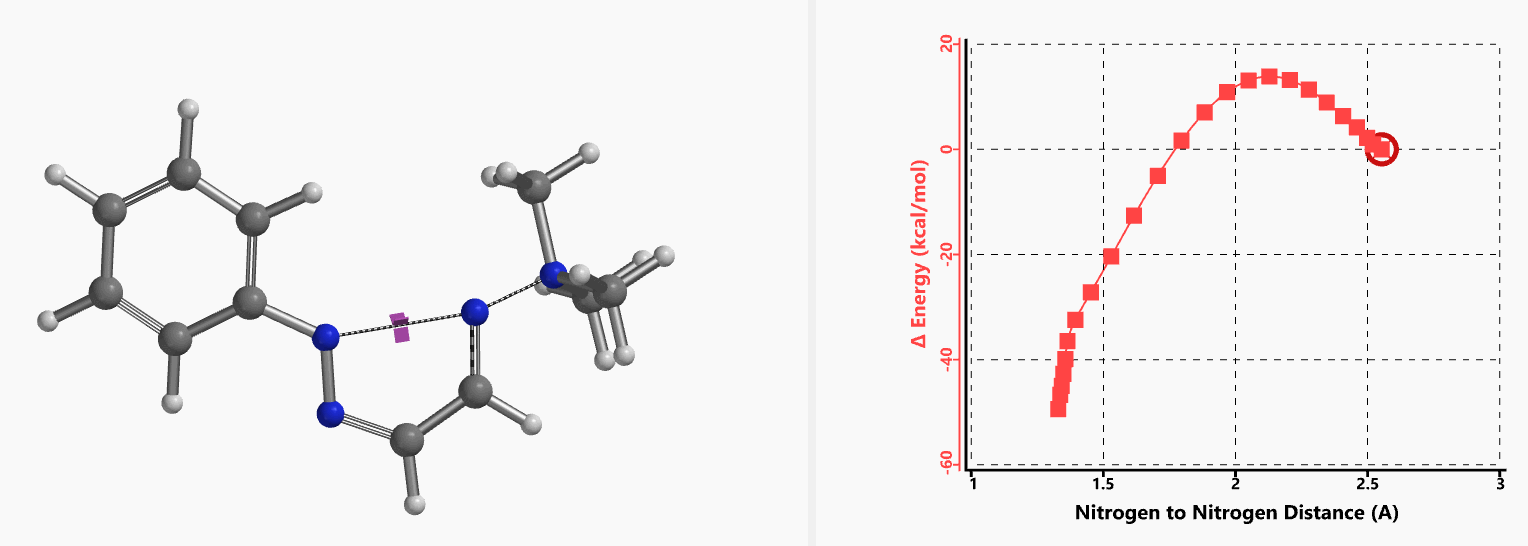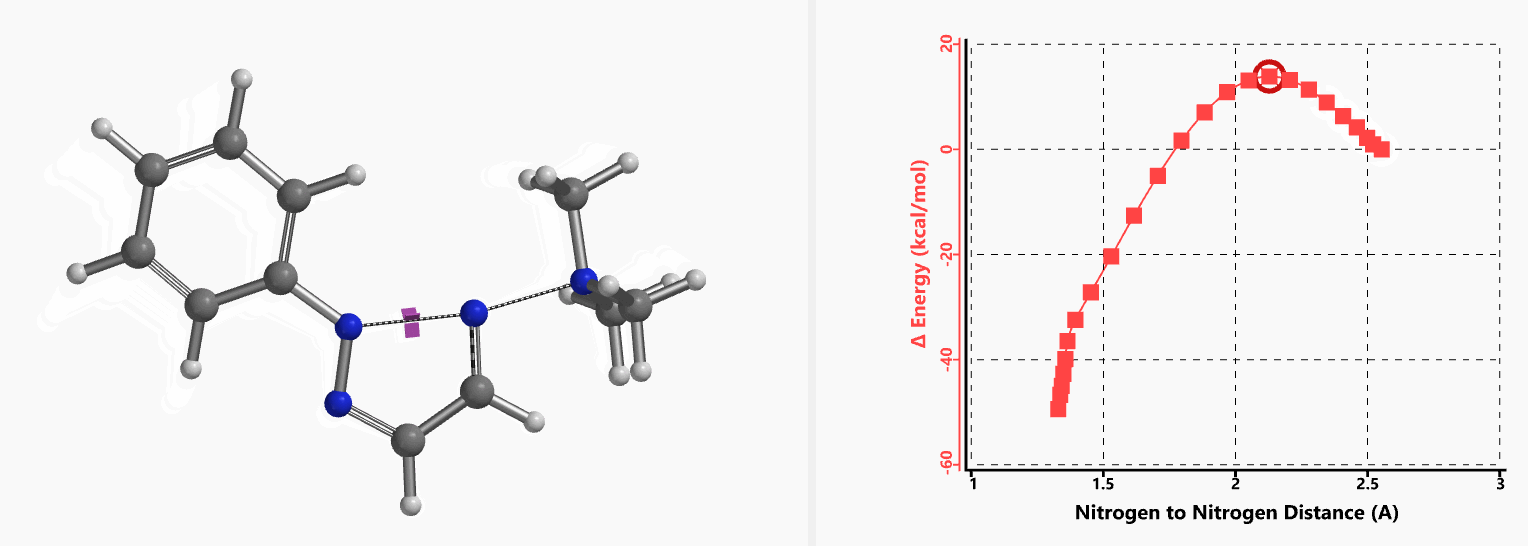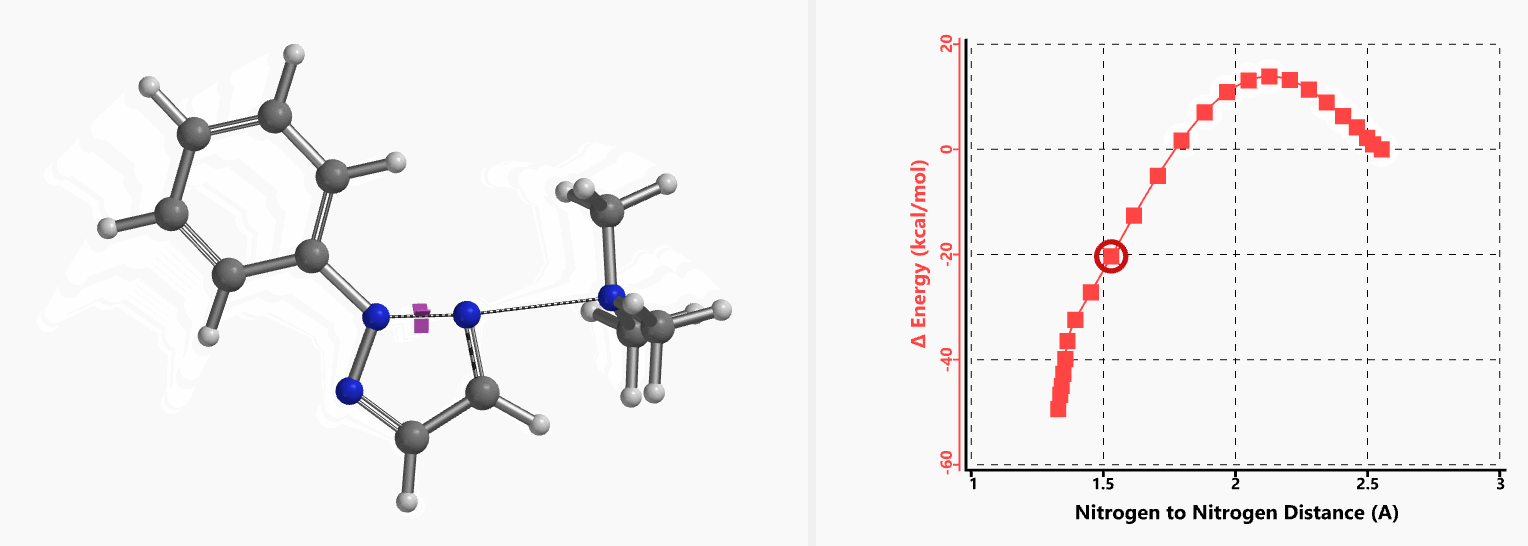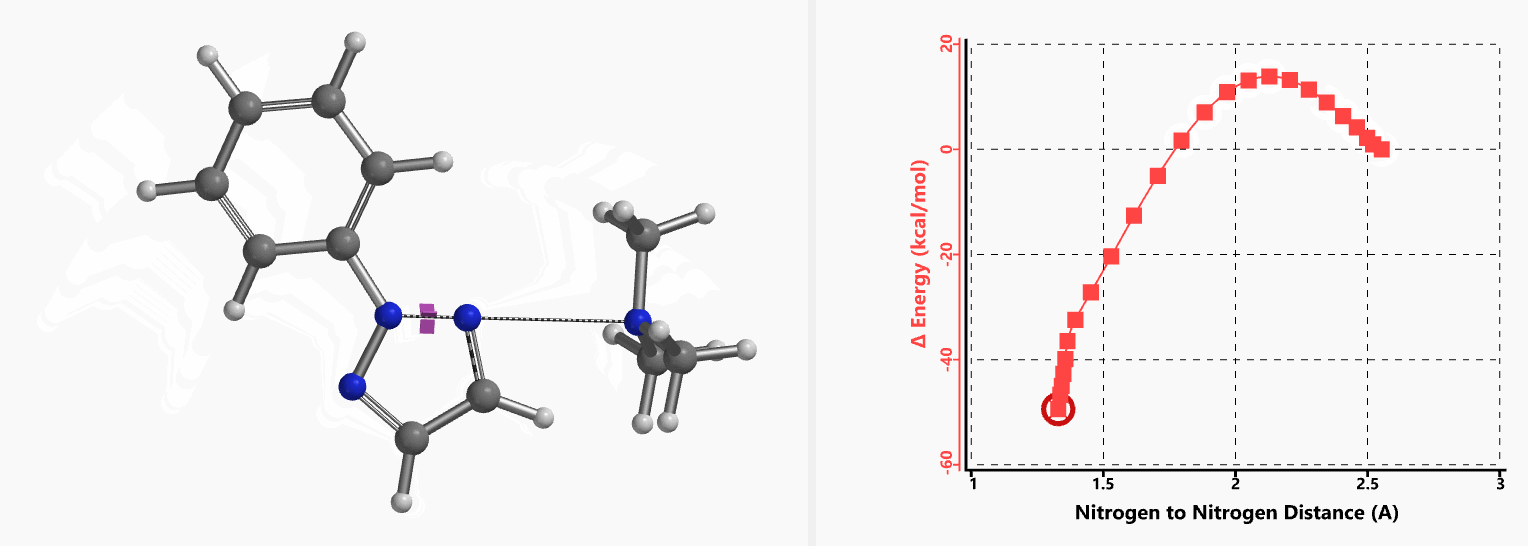
图11. 2-取代-1,2,3-三氮唑合成反应的能量变化曲线
本章节我们通过QM计算验证了sp2中心原子(碳、氮)也可以发生平面内的亲核取代反应,此类转化在经典有机化学中被视为是不可行的[1,7]。QM赋予有机化学家们数据和洞察力,客观地重新评估此类反应的可行性[1,2]。我们从本章学到:
1)sp2中心的SN2反应(SNVσ)(碳、氮原子)是可以发生和预测的;
2)sp2碳反应中心被亲核进攻的可接近性决定了这种 sp2 SN2反应的可行性;
3)逆合成分析时留心使用/发生[4,5]这类反应的可能;
4)定量的QM可行性分析可以为我们提供极好的指导。
接下来,请大家利用氯苯的LUMO、LUMO/Electron Density overlay图 (图12)来解释为什么它不能发生sp2 SN2机制的亲核取代反应,SNAr就可能。
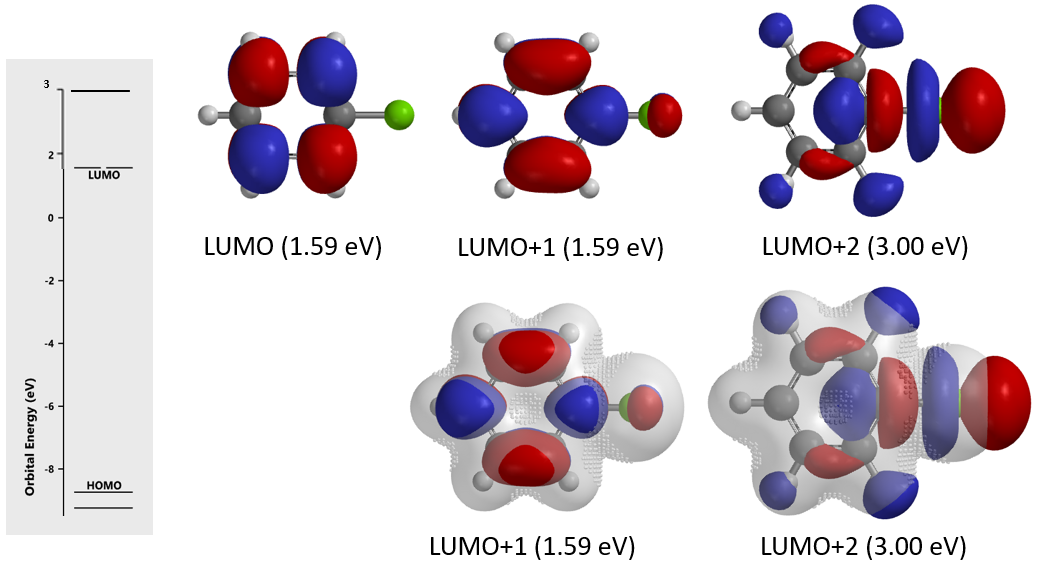
图12. 氯苯的LUMO及LUMO/Electron Density overlay (with inaccessible markers)图
本文由董立亭、赖光华、陈永胜、卫小文编撰。
参考文献:
[1] M. N. Glukhovtsev, A. Pross, L. Radom J. Am. Chem. Soc., 1994, 116, 5961. In-plane SN2 was considered unlikely on the basis of the perception that SN2 attack with inversion at vinylic centers is a high-energy pathway than out-of-plane  -attack. Standard ab initio molecular orbital calculations showed that in-plane σ-type SN2 substitution for Cl- + CH2=CHCl with inversion at unactivated sp2 carbon (136.5 kJ/mol), is actually energetically preferred to the out-of-plane
-attack. Standard ab initio molecular orbital calculations showed that in-plane σ-type SN2 substitution for Cl- + CH2=CHCl with inversion at unactivated sp2 carbon (136.5 kJ/mol), is actually energetically preferred to the out-of-plane 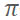 -pathway (178.9 kJ/mol) by 42.4 kJ/mol.
-pathway (178.9 kJ/mol) by 42.4 kJ/mol.
[2] K. Ando, M. Kitamura, K. Miura, K. Narasaka Org. Lett., 2004, 6, 2461.
[3] QM有机化学课堂第二十八章《利用QM对酸催化的非活化烯烃的氢胺甲基化反应机理的探究》。
[4] H. Miyauchi, S. Chiba, K. Fukamizu, K. Andob, K. Narasaka, Tetrahedron, 2007, 63, 5940-5953.
[5] C.Y. Chen, X. Lu, M. C. Holland, S. Lv, X. Ji, W. Liu, J. Liu, D. Depre, P. Westerduin, Eur. J. Org. Chem., 2020, 548.
[6] QM有机化学课堂第三十四章《通过QM预测Chan-Lam反应发生位点》。
[7] J. McMurry, Organic Chemistry, Eighth Edition, Boston, MA, USA: Cengage Learning, 2011; pp 379-380. “Vinylic halides and aryl halides are….unreactive towards SN2 displacement. This lack of reactivity is due to steric factors: the incoming nucleophile would have to approach in the plane of the carbon-carbon double bond and burrow through part of the molecule to carry out a backside displacement.”