 选择语言
选择语言



新闻和活动

关于我们
服务和解决方案
新闻和活动
职业发展
服务平台
QM有机化学课堂
市场活动
新闻
QM有机化学课堂
嘧啶环由于其独特的化学特性而广泛的存在于药物分子中。对于药物化学家来说,以取代的2,4-二氯嘧啶为起始原料,通过芳香亲核取代反应(SNAr)引入嘧啶环是常用的合成策略。而在此过程中我们经常会遇到2 vs 4位氯的选择性取代问题。通常情况下,根据我们的认知,4位的氯要比2位的氯更活泼,更容易被亲核试剂进攻。但在实际的合成工作中,我们也经常会遇到例外[1][2][3],即2位的氯会优先被取代的情况。这里我们通过QM对文献中2,4-二氯嘧啶取代选择性的实例进行了深层次的计算分析,从而找出对应的规律,来指导我们对2,4-二氯嘧啶类化合物取代反应选择性的判断。
图1:C6位是给电子取代基时,2,4-二氯嘧啶亲核取代反应的区域选择性
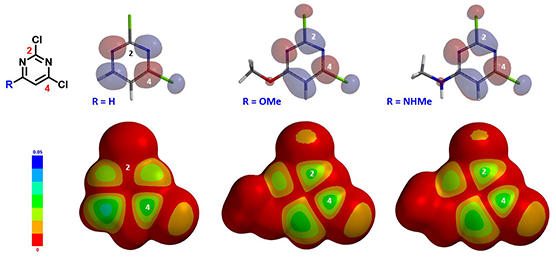
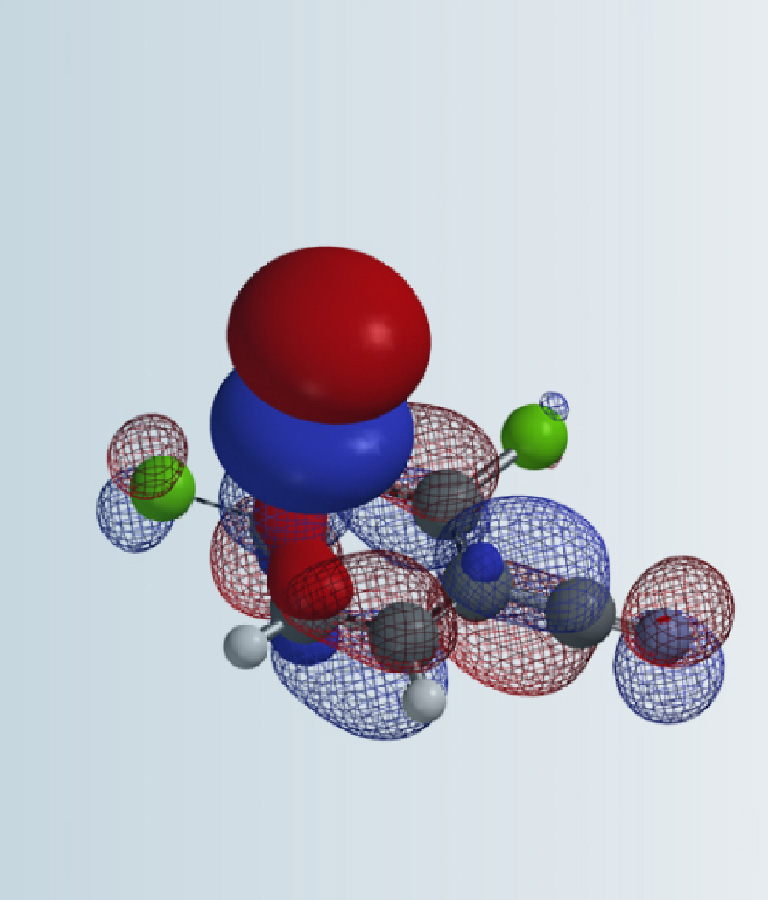
我们用Spartan (DFT wB97X-D/6-31G*) 计算了三种6位取代的2,4-二氯嘧啶的LUMO和LUMO map(图2)。我们可以看到,当6位是氢时,LUMO主要分布在C4位,而C2位几乎没有LUMO分布,这和我们从杂环化学教科书学习得到知识是一致的。而当6位是甲氧基或甲胺基取代时,LUMO有了很大的不同。我们可以看到,LUMO在C2和C4位的分布大小是差不多的,LUMO map的情况也差不多。即根据LUMO和LUMO map分析,当6位是给电子基时,我们可能会得到C2和C4取代的混合物。但前面文献中的实验结果却是高选择性的得到C2取代的产物。也就是说单纯的用LUMO和LOMO map来解释这种反常的取代选择性是不充分的!
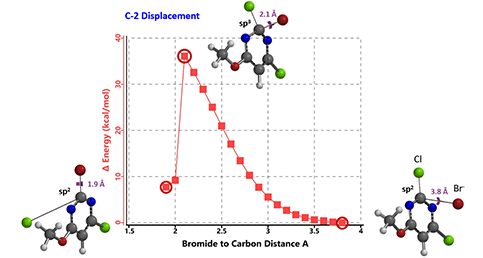
图3:C2取代的反应能量变化曲线(-Br作为替代的亲核试剂)
在这种情况下,我们进一步计算了C2取代反应过程的反应能量变化曲线(图3)。为了简化计算,这里我们用溴来代替亲核试剂,根据我们的经验,这样得到的结果足以支持我们对该过程进行定性分析。C4取代的反应能量变化曲线与此类似(文中未给出)。
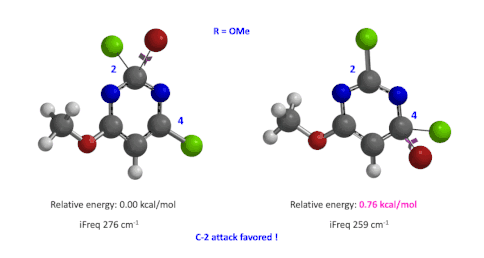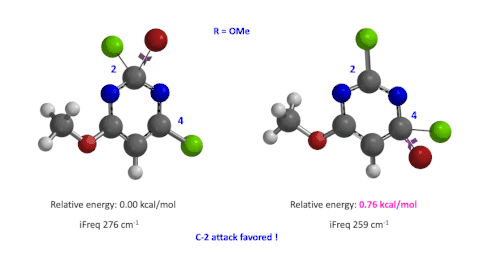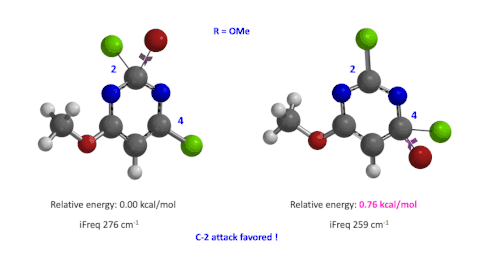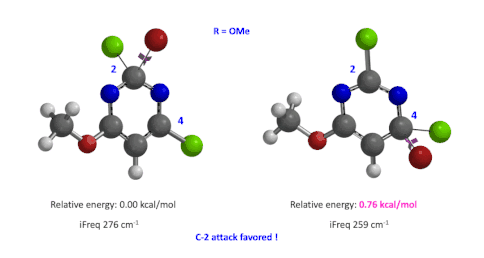
图4:C6位是-OMe时,C2和C4取代的过渡态的相对能量对比
然后,使用这两个过程中能量最高点计算并对比过渡态的能量(图4),我们可以看到,C4要比C2高出0.76 kcal/mol的能量,也就是该反应更倾向于发生在C2位。这与实验结果也是一致的。此外,我们分别计算了两个过渡态的虚频,分别有且只有一个,说明这两个过渡态结构是合理的[4]。
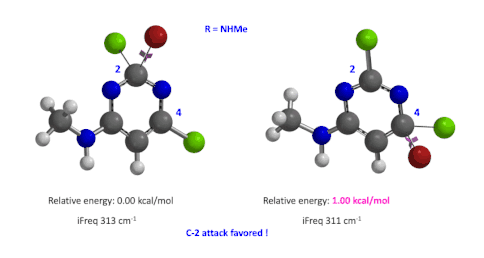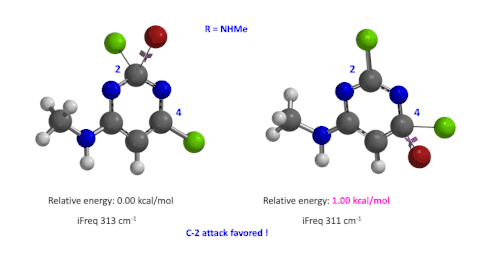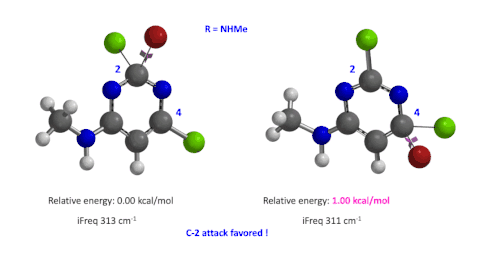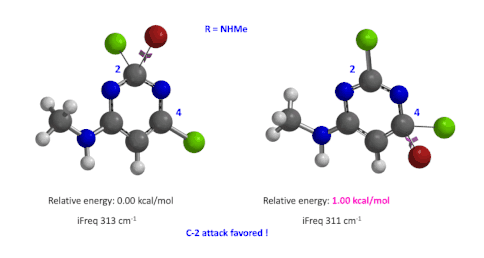
图5:C6位是-NHMe时,C2和C4取代的过渡态的相对能量对比
我们用同样的方法对R为烷基胺取代的二氯嘧啶底物进行了计算和分析,同样,为了简化计算我们将取代基简化为甲胺基,溴作为亲核试剂。我们发现C4位发生取代的过渡态能量比C2位高出1.00 kcal/mol,即反应也是有利于C2进攻的(图5)。
当我们通过分析LUMO来预测两种潜在的竞争反应的区域选择性时,我们是基于哈蒙德假说,即在放热反应中,过渡态的结构更接近于反应物,但这有其局限性。因为,当底物和试剂相互作用时,过渡态中出现了其他干扰因素。我们需要进一步计算和比较反应能量变化曲线和过渡态,以便更接近现实。
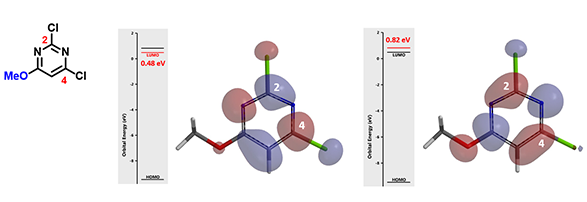
图6:2,4-二氯-6-甲氧基嘧啶的LUMO及LUMO+1
在2,4-二氯-6-甲氧基嘧啶这种情况下(图6),我们可以看到LUMO与LUMO+1之间的能量差是0.34 eV,差值较大,说明LUMO+1参与反应的可能性低,我们在预测其随后的亲核取代反应时是不需要考虑LUMO+1的。

图7:2,4,6-三氯嘧啶的LUMO及LUMO+1
如果我们用氯原子替换6位的甲氧基,三氯嘧啶的LUMO主要分布在C4和C6位,C2位没有LUMO分布,但LUMO和LUMO+1的能量差只有0.25 eV,这里我们就需要同时考虑LUMO+1了,可以看到,C2位有明显的LUMO+1 lobe分布。所以,综合考虑LUMO,LUMO+1,甚至取代的过渡态能量比,我们认为此反应应该得到C4和C2取代的混合物,这与得到区域异构体混合物的实验结果是一致的。
简单总结一下我们从本章及第七章[3]学到的知识,1)对于2,4-二氯嘧啶的取代反应,当5位或6位有强的给电子或吸电子基团时,反应不总是发生在我们常规认知的C4位,很可能会得到C4和C2取代的混合物,甚至只得到C2取代的产物;2)通过LUMO轨道分布预测反应的选择性时,当LUMO和LUMO+1之间的能量差 ≤ 0.25 eV时,需要同时考虑分析LUMO+1;3)C5位是大位阻取代基时,也会影响C4/ C2位取代的选择性[3];4)2,4-二氯嘧啶类化合物参与的亲核取代反应的区域选择性对 C5 和 C6 位取代基的电子及空间位阻效应十分敏感,但该区域选择性很容易通过QM 分析进行合理的解释,并且是高度可预测的。
正文中我们通过QM计算模拟,对2,4-二氯嘧啶类化合物的亲核取代反应的区域选择性进行了分析和解释。接下来,留给大家一个小问题,供您思考。
对于5位是三甲基硅基取代的2,4-二氯嘧啶,其LUMO lobe主要分布在C4位,与LUMO+1的能量差为0.14 eV, 且 LUMO+1 lobe主要分布在C2位。实验结果只观察到C2取代的产物,这是为什么呢?(图8)

图8:2,4-二氯-5-三甲基硅基嘧啶的LUMO及LUMO+1
温馨小提示
如果您有其它精彩的关于嘧啶化学案例,欢迎分享给我们进行QM分析。
参考文献:
[1] L.D. Arnold, H. Maag, W.W. Turner, Jr. WO 2016/168619 A1.
[2] J.M. Large, J.E. Torr, F.I. Raynaud, P.A. Clarke, A. Hayes, F. Stefano, F. Urban, S.J. Shuttleworth, N. Saghir, P. Sheldrake, P. Workman, E. McDonald, Bioorg. Med. Chem. 2011, 19, 836 – 851.
[4] a) Spartan'20 Tutorial and User's Guide (2020). Irvine, CA, USA: Wavefunction, Inc. pp. 158, 442, 459 & 536. b) 本QM章节第二十二及二十四章有关过渡态和虚频的计算。
本文由张人伟、赖光华、董立亭、卫小文编撰。