 选择语言
选择语言



新闻和活动

关于我们
服务和解决方案
新闻和活动
职业发展
服务平台
QM有机化学课堂
市场活动
新闻
QM有机化学课堂
在QM魔法小课堂的第六章《分步多次卤化和偶联反应策略——QM的综合应用》中,我们介绍了一种可以高选择性地在苯胺或苯酚对位溴代的试剂: 四丁基三溴化铵 (TBABr3)。如图1所示,虽然HOMO和碳谱预测胺基邻位反应活性更高,但是使用TBABr3依然可以选择性地得到对位溴代产物[1]。TBABr3独特的对位选择性引起了我们巨大的兴趣,它的反应机理是怎样的呢?让我们利用QM工具来一探究竟吧!
![figure_1[1]-1.png](/cn/Public/Uploads/ueditor/upload/image/20220110/1641801498824372.png)
图1. 化合物A的溴代反应, HOMO和计算的碳谱化学位移
文章报道的TBABr3与苯酚的可能反应机理如图2 (左) 所示[2]。该机理认为Br3阴离子参与反应,拔去酚羟基的质子后,经过一系列的电子转移,得到对位溴代产物。细心的小伙伴可能会发现一个不太合理的地方:富电子Br3阴离子怎么会和同为富电子苯酚直接反应呢?怀着这样的疑问,我们首先计算了Br3阴离子的LUMO (图2右)。它的LUMO轨道能量高达3.07 eV,表明这是一个活性很低的亲电物种,很难发生溴代反应。
![figure_2[1].png](/cn/Public/Uploads/ueditor/upload/image/20220110/1641801530629079.png)
图2. 文献报道机理和Br3阴离子的LUMO能级、能量
同时,我们也计算了该机理下的能量变化曲线(图3)。C–Br键距离从3.8 Å到1.9 Å,这个过程中能量一直增加,找不到一个合理的过渡态,这也说明这种成键方式是行不通的,文献报道的反应机理是不可行的!那么TBABr3还可能以怎样的形式参与反应呢?
![figure_3[1]-1.png](/cn/Public/Uploads/ueditor/upload/image/20220110/1641801562381385.png)
图3. Br3-与苯酚反应中间体结构和能量变化曲线
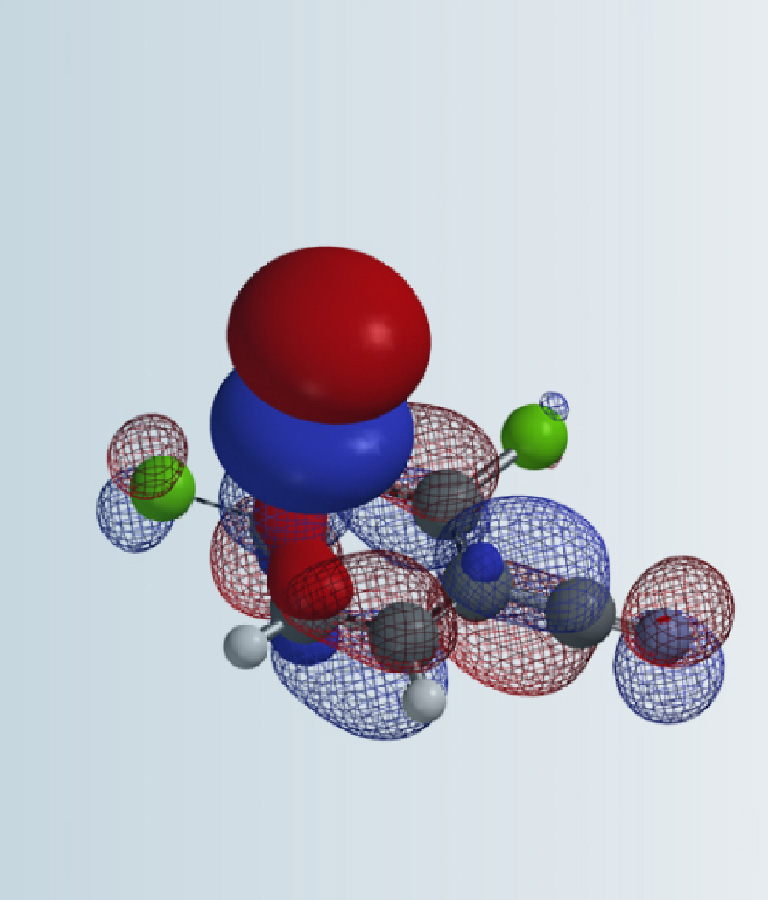
我们猜想四丁基铵正离子不仅仅起到稳定Br3阴离子的作用,很可能作为一个整体参与到了卤代反应。铵基正离子和Br3阴离子存在两种可能的连接方式,为了方便计算,我们将四丁基铵简化为四甲基铵 (TMA)。计算结果显示,TMA+连接在Br-1上的LUMO轨道能量为0.41 eV (图4左)。连接在中间溴原子Br-2上时,计算得到的平衡结构非常独特。TMA+的N位于Br-2和Br-1之间,更接近Br-1 (距离分别为3.96 Å和 4.19 Å),此时相应的 LUMO轨道能量更低,为 -0.34 eV (图4右)。此外, TMA–Br-1–Br-2三元环结构的相对能量比线性的TMA–Br-1低13.42 kcal/mol,所以我们预测它是最可能的卤代反应活性物种。
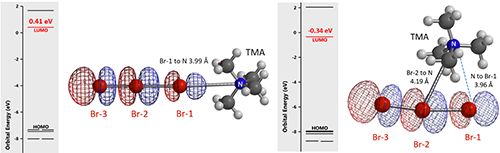
图4. TMABr3的可能的结构,以及它们的LUMO 和 LUMO能量
在寻找反应过渡态的过程中, 为了进行全方位的比较,我们计算了TMABr3与底物之间三种可能的连接方式(图5)。
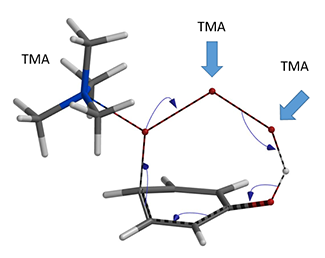
图5. 三种可能的连接方式
计算结果显示,三种不同连接方式的初始结构最终找到了一个同样的过渡态。这也说明TMA+与Br3-的连接是动态的。以TMA+连接在中间溴原子Br-2为例,计算C–Br从3.5 Å到2.2 Å的能量变化曲线 (图6), 在C–Br距离为2.4 Å时,相对能量达到最大值,反应的活化能约为25 kcal/mol。
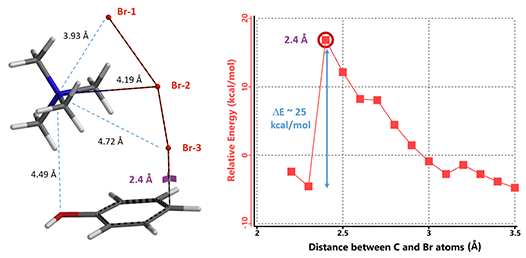
图6. 过渡态计算初始结构示意图,对位取代能量变化曲线
计算得到的反应过渡态中并不存在氢键,这样的过渡态与文献报道的实验结果更加吻合。TBABr3与不存在氢键的二乙基苯胺反应,同样以高选择性得到对位取代产物[3],之前文献中假设的反应机理则不能解释该现象[2]。观察过渡态的结构,可以发现TMA+在空间上非常接近富电子的羟基 (N-O 距离为4.49 Å)。因此我们推想两者存在一定的相互吸引作用,使卤代试剂的Br-3和底物对位C原子的立体电子方向处于最适合发生反应的位置,稳定过渡态,降低活化能。
从过渡态的空间结构来看,溴原子是以接近垂直的角度进攻芳环。我们计算了在接近形成过渡态,C–Br-3距离为2.6 Å时的轨道分布(图7)。卤代试剂以接近垂直的角度进攻芳环,这样可以使两者之间达到最大的轨道匹配度。
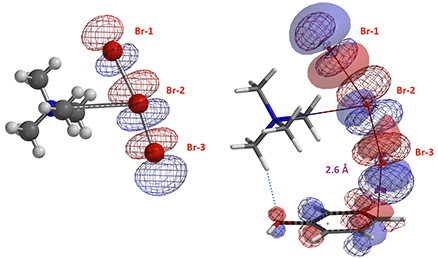
图7. TMABr3 LUMO轨道和C–Br距离2.6 Å时反应结合物中的HOMO (solid) / LUMO (mesh) 轨道示意图
同样C–Br-3距离2.6 Å时的静电势能图显示,四甲基铵正离子上的甲基是非常缺电子的 (图8左, 深蓝色) [4]。电子密度图显示羟基氧和距离最近的甲基上的氢存在非共价相互作用[5],从而形成一个环状的TMABr3 – PhOH反应结合物 (图8右)。
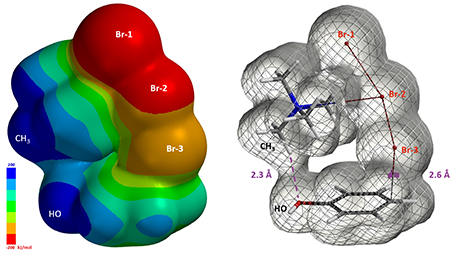
图8. 反应结合物C–Br距离2.6 Å中时的静电势能图 (Isovalue 0.002 e/au3, 99.4%) 和 Electron Density (Isovalue 0.00744 e/au3, 98.0%) 示意图
那么,TBABr3的对位选择性是怎么产生的呢?我们同时对邻位溴代进行了计算,过渡态结构和能量变化曲线如图9所示。邻位溴代反应的活化能约为 45 kcal/mol,比对位卤代的活化能高了大约20 kcal/mol,所以TBABr3可以高选择性地得到对位取代产物。同时,对位溴代时,C–Br键在2.4 Å 达到过渡态,而邻位溴代则是2.1 Å, 这进一步说明反应更倾向于发生在对位。

图9. 对位溴代过渡态结构,邻位溴代过渡态结构和邻位溴代能量变化曲线
本篇小课堂利用QM计算和分析,我们给出了TBABr3卤代反应更加合理的反应机理,揭示了其对位选择性的原因。四丁基铵基阳离子与Br-1和 Br-2形成三元环结构时LUMO能级的能量最低,更可能是反应中真正的活性溴化物种。计算的过渡态结构显示,溴原子以接近垂直的角度进攻芳环,此时可以实现卤化试剂和底物分子轨道的最大重叠。该结构不存在氢键相互作用,与实验结果更加连贯。静电势能图和电子密度图揭示苯酚氧与四甲基铵正离子部分存在非共价相互作用,使反应中心形成适合参与反应的空间关系。邻位溴代反应的活化能比对位卤代时高约20 kcal/mol,这就解释了TBABr3高选择性对位溴代的原因。在QM的帮助下,我们对TBABr3的反应机理有了更加深入准确的理解,希望本文中的探索过程也能给大家更多的启发。
苯酚化合物的对位选择性氯化也已经有多篇报道[6]。其中一种方法是以磺酰氯作为氯化试剂,硫化环戊烷和AlCl3作为催化剂,实现了苯酚的高选择性对位氯化 (图10)[6e]。我们认为AlCl4-硫化环戊烷-Cl一起参与了反应,分别计算了相应对位和邻位氯代的能量变化曲线以及过渡态近似结构的LUMO (图11)。小伙伴们对照上文的内容分析一下它与TBABr3选择性对位溴化有哪些相似之处吧!
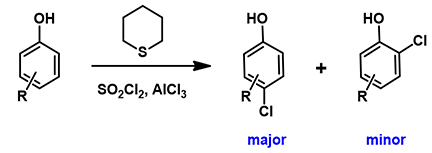
图10. 苯酚化合物的对位选择性氯化反应
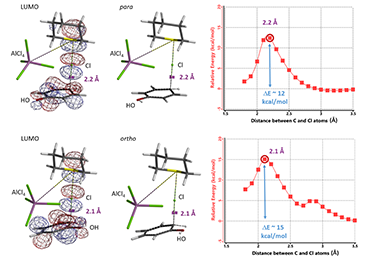
图11. 对位和邻位过渡态结构LUMO以及能量变化曲线
参考文献:
本文由余栋、潘东、沈小莉、赖光华、卫小文编撰。