 选择语言
选择语言



新闻和活动

关于我们
服务和解决方案
新闻和活动
职业发展
服务平台
QM有机化学课堂
市场活动
新闻
QM有机化学课堂
在有机合成中,通过制备缩酮来保护酮是一种常用方法。一般使用乙二醇在酸催化条件下与酮缩合制备缩酮。但是这种方法的缺点是使用大位阻酮制备乙二醇缩酮时,反应温度高、时间久、原料总有剩余,给分离纯化带来困难;另外在缩酮的脱保护过程中,需要高温、强酸条件,很容易导致产物结构遭到破坏。部门的小伙伴们根据文献报道[1]和自己的探索发现了一个易合成、易水解的缩酮体系:Si(OMe)4/TsOH。利用该体系可以高收率地得到易水解的二甲醇缩酮产物。对于这样一个实用的反应,它的反应机理目前还没有人报道过。
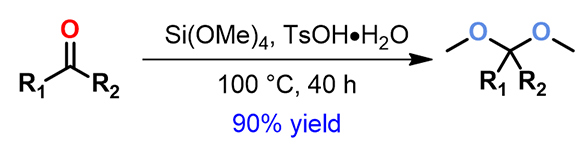
图1. Si(OMe)4/TsOH体系合成二甲醇缩酮
接下来,我们将会利用QM计算来探索它的反应机理。
如图2所示,我们根据已有的化学知识给出了如下的反应机理。首先,酮和四甲氧基硅分子通过类似Wittig反应的四元环过渡态 I 将一个甲氧基转移到羰基碳上,得到中间体 II 。在质子作用下C-O键断裂,形成氧鎓正离子中间体 IV 和(MeO)3SiOH。接下来第二个甲氧基再次转移到羰基碳上得到缩酮产物C和硅酸二甲酯D。这个机理是否合理呢?我们可以通过QM计算相应的过渡态和活化能来检验每一步是否可行。
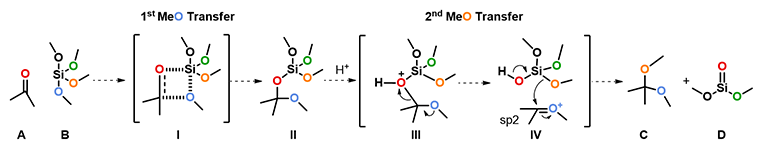
图2. 反应可能的机理
根据QM魔法小课堂第九章的内容,我们模拟酮和四甲氧基硅分子经由四元环过渡态过程将第一个甲氧基转移到羰基碳上的能量变化曲线(图3)[2]。Si-O之间的距离,从彼此远离的3.6 Å逐步接近至进入共价键作用范围的1.7 Å,C-O之间的距离从3.3 Å到1.4 Å,步长0.1 Å。活化能曲线的能量最高点近似为反应的过渡态,其结构如图3(中)所示,活化能为20.7 kcal/mol,是一个合理的活化能数值[3],所以第一个甲氧基的转移过程可以通过该过渡态发生[4]。
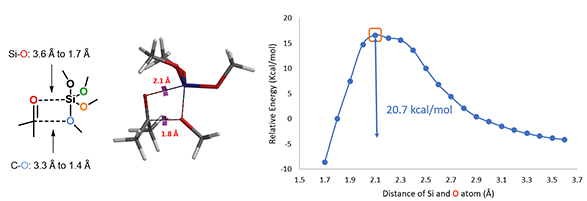
图3. 第一个甲氧基转移的过渡态、能量变化曲线
通过第一个甲氧基转移,我们会得到中间体 II ,接下来反应是否可以通过O质子化的中间体 III 进行呢(图4左)?通过计算(MeO)3SiOH和MeOH的静电势图(图4右),我们可以非常清楚的看到(MeO)3SiOH的酸性更强,(MeO)3SiO相对于MeO是一个更好的离去基团,所以反应可以通过O质子化的中间体 III 进行[5]。
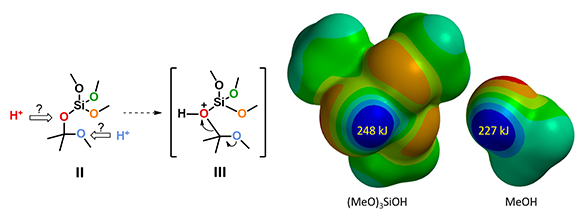
图4. (MeO)3SiOH和MeOH的静电势图
如图5所示,接下来就是甲醇进攻来实现第二个甲氧基的转移。但是,甲醇是哪里来的呢?是不是来自于(MeO)3SiOH的分解呢?
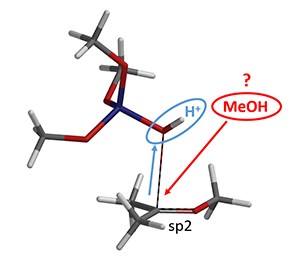
图5. 甲醇进攻示意图
活化能的计算结果告诉我们这一步并没有这么简单。从图6的能量变化曲线我们可以看到,如果(MeO)3SiOH想要分解生成甲醇,活化能为64 kcal/mol。这是一个非常大的能垒,这样的过程一般不能发生[3]。
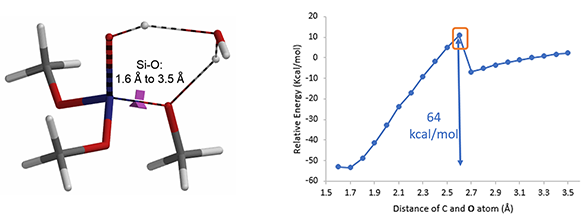
图6. (MeO)3SiOH的分解生成甲醇的过渡态、能量变化曲线
那么甲醇还可以来自于哪里呢?我们将目光聚集到了反应过程中使用的催化剂上,我们使用的是吸湿性的一水合对甲苯磺酸,水分促进了Si(OMe)4分解生成甲醇和SiO2(图7)。
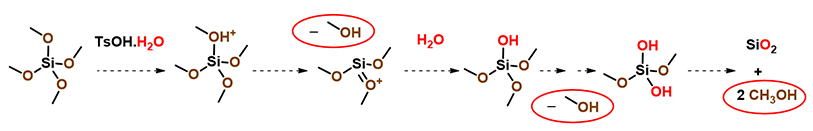
图7. TsOH-H2O与四甲氧基硅反应生成甲醇
解决了MeOH的来源,我们再次来到第二个甲氧基转移的过程。质子化后的中间体 III 可以通过两个可能的机理实现第二个甲氧基的转移:1. 直接通过协同的过程接受甲醇的进攻。2. C-O键断裂,形成的氧鎓离子中间体接受甲醇的进攻。这里我们计算了通过协同机理进行第二个甲氧基转移的活化能。如图8所示,活化能只有8.93 kcal/mol,这是一个非常低的能垒,表明这样一个过程可以顺利发生[3]。由于我们找到了一个非常合理的协同过渡态,而第二种机理则需要形成一个非常高能量的氧鎓离子中间体,这里就没有进一步计算。
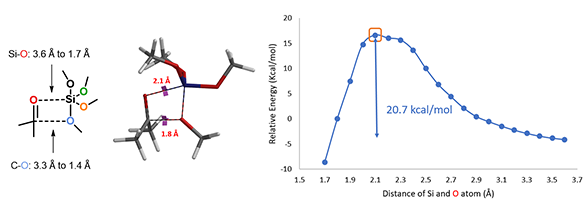
图8. 第二个甲氧基通过协同机理转移的过渡态、能量变化曲线
根据计算结果我们修正了最初假设的机理(图9)。首先,酮和四甲氧基硅分子通过四元环过渡态 I 实现第一个甲氧基的转移,得到中间体 II ,活化能约为20 kcal/mol,这一步不需要酸催化的参与。中间体 II 中C-O-Si的氧原子被TsOH质子化后,通过协同机理,经由过渡态 III 实现第二甲氧基的转移,活化能约为9 kcal/mol,最终得到了产物缩酮C。一水合对甲苯磺酸在这里有两个作用:一是催化第二个甲氧基的转移;二是水分促进四甲氧基硅水解得到反应需要的甲醇。
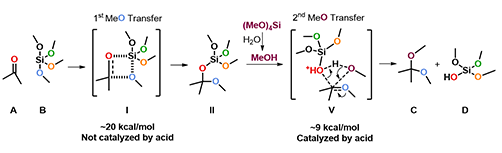
图9. 根据计算结果修正后的可能机
由于使用四甲氧基硅烷合成缩酮的方法还没有相关的文献报道,下面向大家分享具体的实验步骤:
在氮气保护条件下,向反应底物酮 1 (1.0 mmol, 1.0 eq.)中加入2.5 mL Si(OMe)4以及TsOH•H2O (0.04 mmol, 0.4 eq.)。反应在100 ℃条件下搅拌40 h。反应结束后冷却至室温,加入适量的乙酸乙酯稀释,饱和食盐水洗涤3次,有机相干燥、浓缩,进行硅胶柱层色谱分离得到二甲醇缩酮产物。
此外,二甲醇缩酮产物非常容易水解,可以在非常温和的条件下(TFA/CHCl3/H2O, rt, 1 h)快速水解为酮,对一些在强酸或者加热条件下稳定性较差的底物非常适用。如果大家在工作中需要合成缩酮,非常推荐大家来尝试一下Si(OMe)4/TsOH的方法哟。
温馨小提示:
下一章的QM魔法小课堂将会为大家介绍,当HOMO lobe大小和碳谱数值都很接近时怎样预测亲电卤代的区域选择性,敬请期待!
本文由潘东、周正权、刘文锋、石谷沁、董立亭、赖光华、卫小文编撰。
参考文献:
[1] a) Nasarow et al. Zhurnal Obshchei Khimii, 1959, 29, 106. b) Sakurai, H. and Love, J.A., "Silicon(IV) Methoxide" in Encyclopedia of Reagents for Organic Synthesis, 2009, John Wiley & Sons.
[2] Spartan’18 Tutorial and User’s Guide (2019). Irvine, CA, USA: Wavefunction, Inc.
[3] 活化能小于10 kcal/mol的反应通常在低温至室温的条件下就可以进行。活化能在10-20 kcal/mol之间,反应一般需要加热。大于20 kcal/mol,需要加热和更长反应时间,大于30 kcal/mol则需要更高的反应温度。活化能大于40 kcal/mol,反应则一般很难发生。
[4] 利用氧原子质子化后的酮计算能量变化曲线没有成功。
[5] 这一步反应可以通过质子化的中间体 III 进行是因为(MeO)3SiOH相比于MeOH是一个更好的离去基团。但是从碱性方面考虑,MeO中的氧应该更容易被质子化,因为它的共轭酸酸性更弱。就像缩醛胺在酸性条件下生成亚胺,虽然RNH2具有更强的碱性会优先质子化,但是H2O是一个更好的离去基团。