

新闻和活动

关于我们
服务和解决方案
新闻和活动
职业发展
服务平台
QM有机化学课堂
市场活动
新闻
QM有机化学课堂
在上一章的理论课里,我们提到了利用哈蒙德假说,可以构建反应过渡态的结构,计算反应活化能,辅助预测化学反应。小伙伴们是不是都很好奇,想在实际工作中演练一下呢?
我们来看下面这个例子:化合物 1 的烷基化反应(图 1),该烷基化反应会得到的是 1 位还是 2 位的烷基化产物呢?如果得到的是混合物,两者的比例又会怎么样呢?
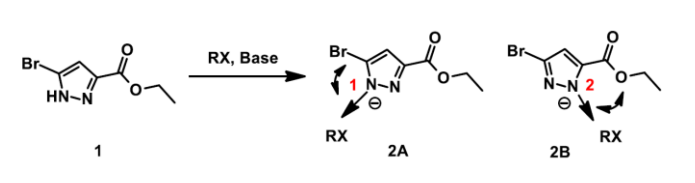
图 1. 吡唑 N 烷基化的区域选择性
带着这两个问题,我们首先分析一下该反应的进程:碱性条件下,吡唑 NH 去质子转化为 N 负离子,之后发生烷基化。
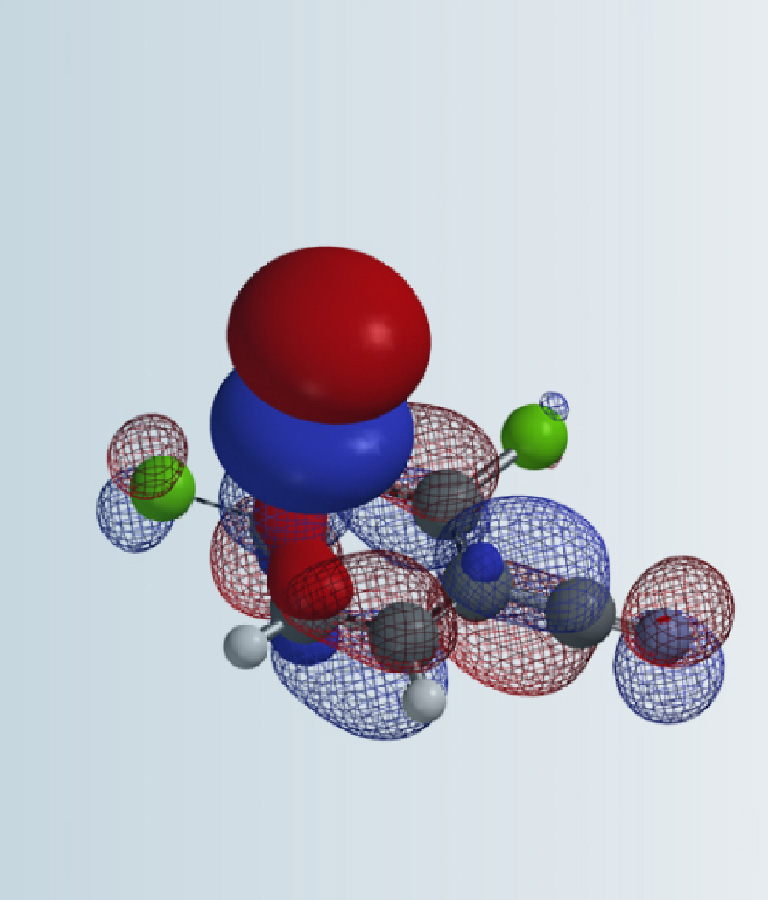
利用本书第三章学习的静电势图,可以比较吡唑的两个异构体中,NH 的酸性强度,判断哪个 NH 会优先和碱反应形成负离子(图 2 A 和 B)。异构体 A 中的 NH 静电势(281 kJ/mol)略高于异构体 B 中的 NH 静电势(266 kJ/mol),但差异并不显著;而且在这个案例中这点也并无太大意义,因为 NH 被拔氢后,负电荷在两个氮原子上是离域的,因此负离子状态下,两个 N 原子上的 HOMO lobe 大小也非常接近(图 2 C)。
所以,在这个案例中,仅凭静电势图和 HOMO 分布难以判断反应的选择性。同时,由于两个氮原子的邻位分别有不同的取代基,会带来不一样的电子效应和位阻效应。因此我们需要进一步分析,来预测产物情况。
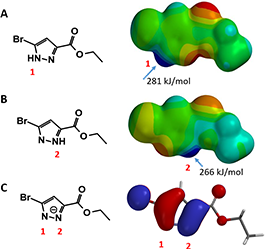
图 2. A 和 B:化合物 1 两种异构体的静电势图;C:化合物 1 被拔氢后负离子的 HOMO 示意图
现在,有请本章节的主人公——活化能计算(Reaction Energy Profile),隆重登场!
活化能计算的基本概念是通过 QM 模拟反应成键或断键的过程,寻找反应过渡态,得到反应的活化能值,利用活化能的大小判断反应的难易,并利用活化能差值计算出不同产物的比例。
再来看我们的例子,吡唑化合物 1 的烷基化反应为典型的 SN2 反应,其机理为 N 负离子逐渐接近卤代烷烃,形成 C-N 键,同时发生 C-X 键的断裂。
以CH3Br为反应物,我们分别在CH3Br的C原子与化合物 1 的 N1 或 N2 原子之间模拟 C-N 键的形成,即定义 C-N 之间的距离,从彼此无相互作用的远距离 3.8 Å,逐渐接近至进入共价键作用范围的 1.9 Å,计算这一过程中,系统能量的变化(图 3)。
我们发现 C-N 原子间的距离缩短至 3.0 Å 时,系统能量开始上升,说明反应物分子开始相互作用。键长缩短至 2.1 Å 左右时,能量达到最高点,反应物分子形成了过渡态络合物。之后 C-N 之间逐渐形成稳定的共价键,C-Br 键断裂,系统能量降低,烷基化反应趋于完毕。
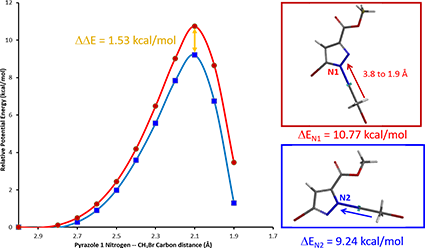
图 3. N1(红)、N2(蓝)活化能以及二者的活化能差值
通过计算,我们发现 N1 位置发生烷基化反应所需的活化能约为 10.77 kcal/mol,N2位置发生反应的活化能约为 9.24 kcal/mol(图 3)。N1 位烷基化所需要的活化能比 N2 位的要高,说明反应更容易发生在 N2 位上。
根据两个反应活化能的差值,我们可以进一步利用阿伦尼乌斯公式(图 4)定量计算两个产物的比例(具体推导可详见本书第八章《站在巨人的肩膀上:哈蒙德假说》)。
根据图 3,N1 与 N2 位烷基化反应的活化能差值 ∆

