 Choose language
Choose language
< Return to main menu
Choose language



News & Events

Services & Solutions
News & Events
Career
Platform
Magical Power of Quantum Mechanics
Events
News
Magical Power of Quantum Mechanics
Ying Lu, Wenfeng Liu, Qiuyue Wang, Yongsheng Chen, John S. Wai
Regioselectivity for SnAr reactions of halo pyrimidines is sensitive to the presence of other substituents on the ring. Intuitively, they are not easy to understand, yet fully accountable for with proper QM analyses (See QM chapters 1, 7, 29). In this chapter, we’ll discuss the longstanding puzzle of dichotomy in regioselectivity observed for SnAr reactions with 2-MeSO2-4-chloropyrimidine. Similar to 2,4-dichloropyrimidine, SnAr reactions of 2-MeSO2-4-chloropyrimidine with amines and Stille coupling occur selectively at C-4, but substitution reactions with alkoxides and formamide anions occur selectively at C-2 (Figure 1).

Figure 1. Dichotomy in regioselectivity of various reaction types with 2-MeSO2-4-chloropyrimidine
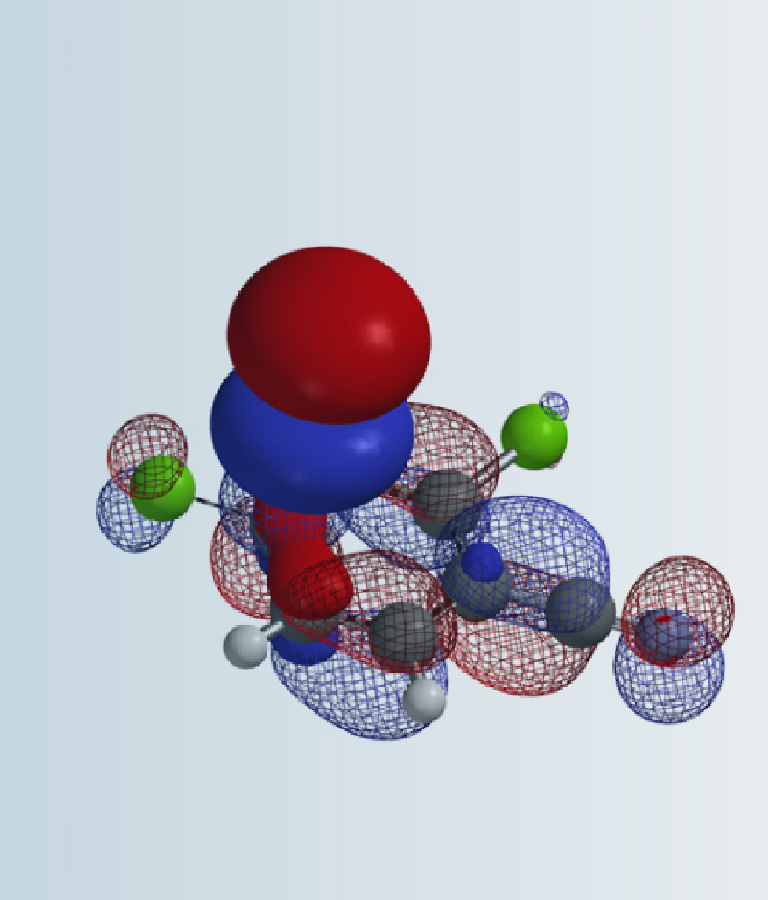
In 1993, SnAr of 2-MeSO2-4-chloropyrimidine with alkoxide was reported to be exclusively at C-2 and proceed at surprisingly low temperature (-78°C)[1]. Even though this unique selectivity has been utilized quite often in MedChem syntheses[2] , there is no further analysis on why it is C-2 selective. Below are results from our QM analyses. LUMO (-0.23 eV) calculated for the pyrimidine shows a lobe centered on C4. LUMO+1 (-0.14 eV) is only 0.09 eV higher in energy and has a lobe centered on C-2 (Figure 2). Together, they suggest that we could have a mixture of C-4 and C-2 substitution products. Obviously, the above LUMO analysis, based on the equilibrium geometry of the pyrimidine, fails to account for the high C-2 selectivity observed.
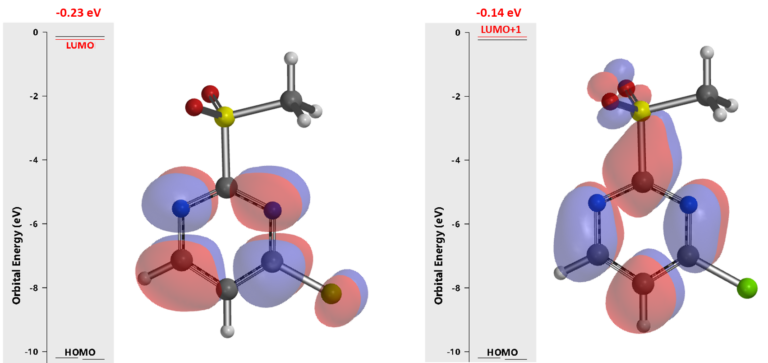
Figure 2. LUMO and LUMO+1 of 2-MeSO2-4-chloropyrimidine
We reasoned that 2-MeSO2-4-Cl-pyrimidine and alkoxide form a hydrogen bond complex. Equilibrium geometry calculated for the complex shows a distance of 1.86 Å between the alkoxide oxygen and the methyl hydrogen of the MeSO2 group (Figure 3, left). There is significant electron density between the oxygen and hydrogen atoms (Figure 3, middle and right), supportive of our hypothesis. It is likely that this is a key factor in directing the SnAr reaction to proceed at the C-2 vs C4 positions.
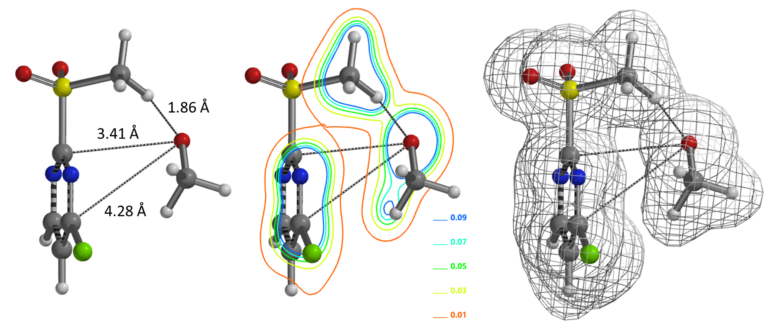
Figure 3. Equilibrium geometry, ED contour and density maps of pyrimidine-alkoxide HB complex
(Calculation level: wB97X-D/6-31+G*)
Next, we modeled the SnAr at C-2 with C-2 to O distance changing from 3.41 Å to 1.51 Å with step size of 0.05 Å. Calculated energy barrier is about 0.25 kcal/mol, accounting for the -78°C low reaction temperature (Figure 4, left hand side). The hydrogen bond between MeSO2 H and alkoxide oxygen in the hydrogen bonded complex is maintained, stabilizing the reaction transition state. There is no energy barrier for subsequent elimination of the MeSO2 group (not shown) to form the C-2 substitution product.
Similarly, calculation of the reaction energy profile for the C-4 substitution (Figure 4, right hand side) shows an energy barrier of ~4.11 kcal/mol, 3.86 kcal/mol higher than that at the C-2 position, accounting for why the reaction occurs exclusively at the C2 position. The higher barrier corresponds to energy cost in breaking of the MeSO2 H and alkoxide oxygen hydrogen bond.
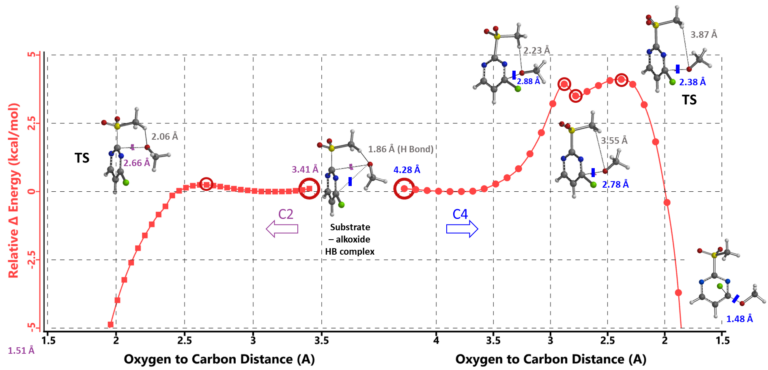
Figure 4. Reaction Energy Profiles of SnAr reaction between 2-MeSO2-4-Cl-pyrimidine and alkoxide: C-2 vs C-4
Subsequent transition state geometry calculation reveals a relatively long carbon to oxygen distance of 2.64 A, single and relatively low iFreq of 88 cm-1 for SnAr at C-2, suggestive of a relative early transition state. The C-4 transition state is 3.87 kcal/mol higher in energy (Figure 5)[3], in accordance with results from REP calculations above.
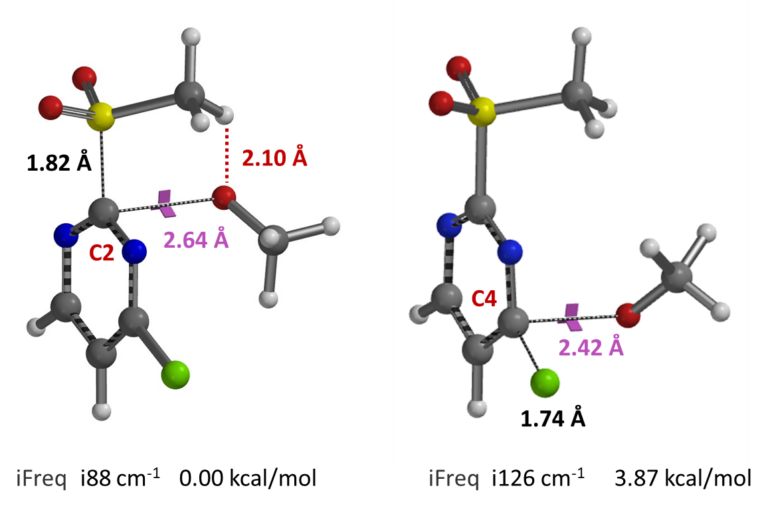
Figure 5. Transition state geometries and imaginary frequencies for SnAr at C-2 and C-4 positions
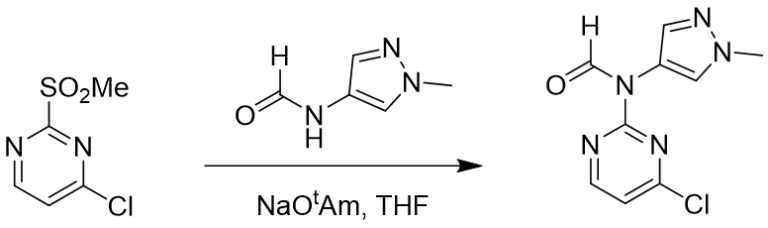
Figure 6. Reaction with aromatic formamide compound
SnAr reaction between 2-MeSO2-4-Cl-pyrimidine and aromatic formamide also occurs selectively at C-2 position [4,5]. Based on what we have learned above, we first calculated for the equilibrium geometry of the pyrimidine and methylpyrazole formamide anion hydrogen bond complex (Figure 7). Results are supportive of the presence of a hydrogen bond (2.24 Å) between the H of the MeSO2 group and the N of the formamide (Figure 7, left and middle) and reveal the presence of a pi-pi interaction between the two aromatic rings (Figure 7, right).
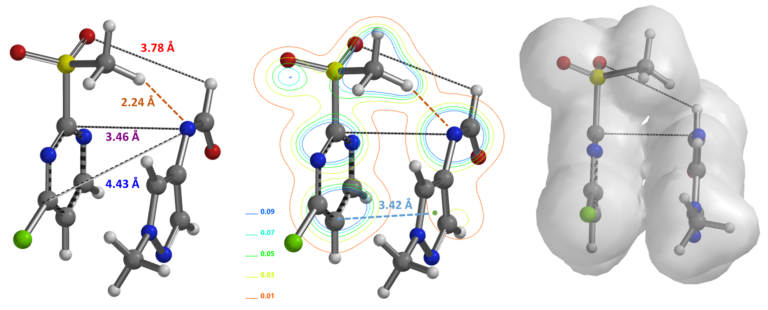
Figure 7. Equilibrium geometry, ED contour and density maps of pyrimidine-formamide anion complex
Modeling of the SnAr reaction at C-2 provided a calculated energy barrier of ~8.75 kcal/mol, consistent with reported reaction temperature of 0°C. The hydrogen bond between MeSO2 H and formamide N in the hydrogen bonded complex is preserved in transition state (Figure 8, left hand side). Similarly, calculation of the reaction energy profile for C-4 substitution (Figure 8, right hand side) shows a higher energy barrier of ~14.71 kcal/mol resulting from the loss of the hydrogen bond. The difference in energy barrier of 5.96 kcal/mol accounts for the highly selective C-2 substitution observed.
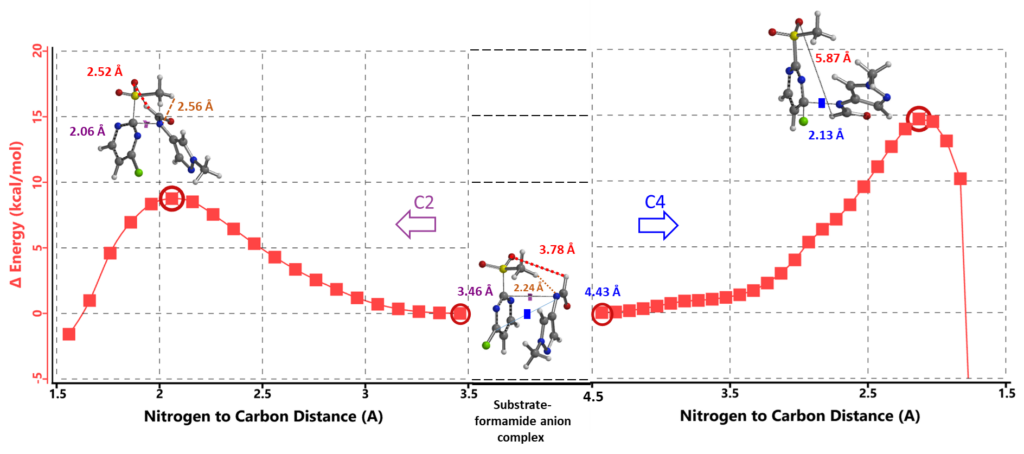
Figure 8. REP of SnAr reaction between 2-MeSO2-4-Cl-pyrimidine and formamide anion: C-2 vs C-4
Shown in figure 9 are the calculated transition state geometries and each of their single imaginary frequencies. Energy of the C-4 transition state is 4.12 kcal/mol higher than that at the C-2 position.
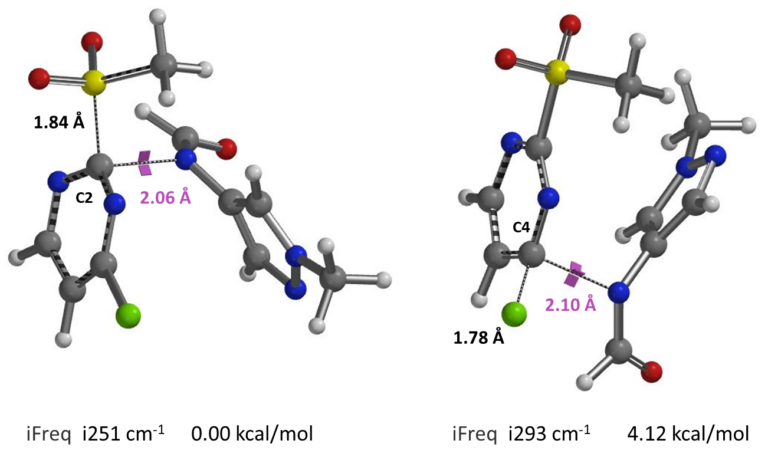
Figure 9. Transition state geometries and iFrequencies calculated for SnAr reaction at C-2 & C-4 positions
The formyl group enables deprotonation of the formamide under mild conditions. Electron density contour maps of the C-2 transition state structure (Figure 10) reveal two important interactions that lead to the high C-2 regioselectivity observed for the SnAr reaction:
1. Hydrogen bond (2.58 Å) between SO2CH3 H and formamide anion N, similar to the alkoxide discussed above; 2. A second hydrogen bond formed between the formamide H and one of the two MeSO2 oxygens (2.53 Å), further lowering its energy. Together, they account better for why the formyl group is so crucial for the high efficiency and selectivity observed for the reaction.
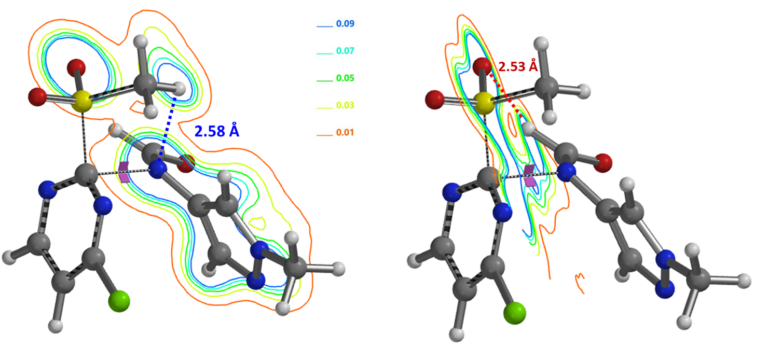
Figure 10. Electron density contour maps of the C-2 transition state revealing two hydrogen bonds
From QM analyses for the two reactions above, we could see why they are highly C-2 selective
1. Non-covalent interaction between the alkoxide/formamide anion with the acidic proton of the methanesulfonyl group, forming hydrogen bond complexes;
2. The nucleophiles need to be anionic for formation of the hydrogen bond complexes;
3. The hydrogen bonds direct the nucleophile to attack at C-2. They are preserved in the transition state, lowering their energies relative to C-4 attack.
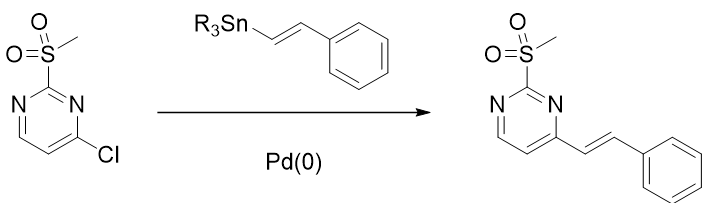
Figure 11. C-4 selectivity for Stille reaction
So why does the oxidative addition reaction occur at the C4 position? Recently, Chatelain [6] et al. reported that methylsulfones display intermediate level of reactivity between that of nitroarenes and aryl halides in Pd(0)-catalyzed Suzuki cross coupling reaction (Figure 12), with trifluoromethyl sulfone group providing the best conversion. Similarly, this reported trend in reactivity could account for faster oxidative addition at C-Cl vs C-O2SMe bonds, accounting for the C-4 selectivity observed with the Stille coupling reaction.
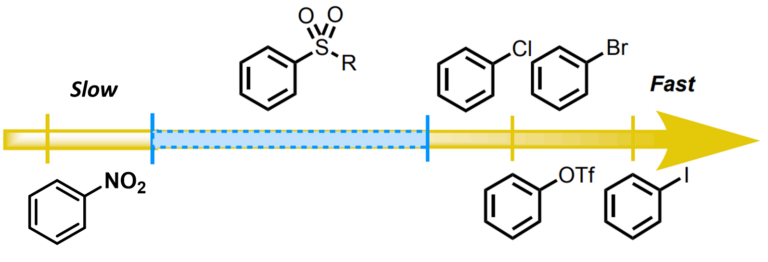
Figure 12. Relative reactivity of aryl electrophiles in Suzuki cross coupling reaction
2-MeSO2-4-Cl-pyrimidine forms hydrogen bond complexes with alkoxide and formamide anion via its acidic MeSO2 proton. This hydrogen bond directs the SnAr reaction at C-2 and lowers the energy of the transition state. On the other hand, C-4 attack will require breaking of the hydrogen bonding, increasing the energy barrier. When the reaction involves hydrogen bond, non-covalent interaction (NCI)[8], steric or other electronic interactions, etc. (see chapters 9, 23, 26, 33, 42), their relative contributions are intuitively difficult to compare. QM calculations enable us to evaluate them in a more objective manner. In the process, we learn to go beyond knowing reactions of interest to understanding them better. It is crucial that we build our computational models based on experimental conditions and observations. If you come across other interesting chemistry, please let us know, let’s analyze and learn together.
The reaction of 2-MeSO2-4-Cl-pyrimidine with secondary amine only occurs selective at C-4. How to rationalize it?
Figure 13. Reaction of 2-MeSO2-4-Cl-pyrimidine with secondary amine
Secondary amines, with lone pair electrons and hydrogen on the N atom, have various potential ways to interact with the MeSO2 group and sp2 N of the pyrimidine. Calculation shows that they form a complex with a ΔE of -7.61 kcal/mol (structure shown in Figure 14). Electron density contour map reveals:
1. Strong non-covalent interaction (2.98 Å) between the secondary amine nitrogen (lone pair electrons) and the electrophilic pyrimidine C-2 carbon.
2. A hydrogen bond (2.55 Å) between the amine nitrogen and MeSO2 group hydrogen; complex with the structure shown is lower in energy (> 1 kcal/mol) than other ones being evaluated in which the amine H hydrogen bond to the MeSO2 oxygen.
3. Two charge-charge interactions between the hydrogens on the secondary amine methyl groups and the pyrimidine N-1 and N-3 (Natural charges).
How would you analyze this reaction further? [9]
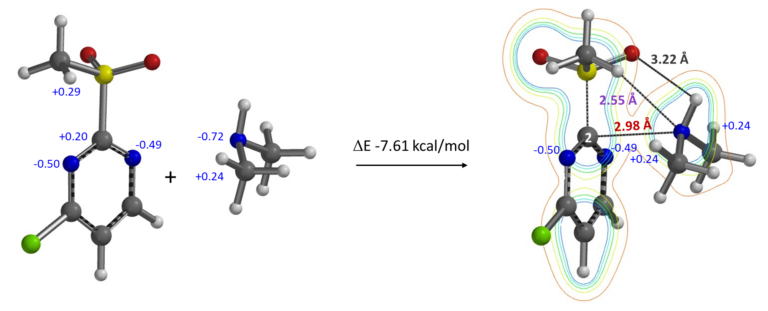
Figure 14. Formation of 2-MeSO2-4-Cl-pyrimidine-secondary amine complex. Electron density contour map and natural charges (blue marker) of the resultant complex
References:
[1] M. L. Falck-Pedersen, B. Tore, U. Kjell, Acta Chem. Scand. 1993, 47, 72.
[2] C. M. Serrano, R. E. Looper, Org. Lett. 2011, 13, 5000; A. S. Carlson, H. R. Cui, A. Divakaran, J. A. Johnson, R. M. Brunner, W. C. K. Pomerantz, J. J. Topczewski, ACS Med. Chem. Lett. 2019, 10, 1296.
[3] F. M. Bickelhaupt, K. N. Houk, Angew. Chem. Int. Ed. 2017, 56, 10070. Small HOMO and LUMO energy gap and little structure distortion from reactant to transition state leads to low activation energy.
[4] C. M. Li, F. Haeffner, S. J. Wang, C. C. Yuan, D. Shang, X. L. Shi, B. Ma, B. T. Hopkins, E. M. O’Brien, Org. Process Res. Dev. 2022, 26, 137.
[5] B. Barlaam, R. Ducray, C. Lambert-van der Brempt, P. Plé, C. Bardelle, N. Brooks, T. Coleman, D. Cross, J. G. Kettle, J. Read, Bioorg. Med. Chem. Lett. 2011, 21, 2207.
[6] P. Chatelain, A. Sau, C. N. Rowley, J. Moran, Angew. Chem. Int. Ed. 2019, 58, 14959.
[7] B. L. Mylari, P. J. Oates, W. J. Zembrowski, D. A. Beebe, E. L. Conn, J. B. Coutcher, M. T. O’Gorman, M. C. Linhares, G. J. Withbroe, J. Med. Chem. 2002, 45, 4398.
[8] QM Chapter 42 https://wuxibiology.com/visualizing-hydrogen-bonds-using-electron-density-maps/
[9] To continue with the analysis, we’ll use the (global minimum energy) structure obtained for 2-MeSO2-4-Cl-pyrimidine-secondary amine complex (shown in figure 14) to set up C-2 and C-4 SnAr REP calculations. Then it will become obvious that the energy barrier of the C-4 substitution is lower than that for C-2. To understand why, transition state calculation, followed by activation strain (distortion) analyses[3] will be very informative. They reveal that the C2- transition state is associated with a larger strain energy, the key reason why SnAr with M. W. Crowder, Bioorg. Med. Chem. Lett., 2013, 23, 5855