 Choose language
Choose language
< Return to main menu
Choose language



News & Events

Services & Solutions
News & Events
Career
Platform
Magical Power of Quantum Mechanics
Events
News
Magical Power of Quantum Mechanics
Introducing fluorine into organic compounds can significantly alter their physical and biological properties, by changing their conformation, non-covalent interaction with target proteins, lipophilicity, acidity/basicity, metabolic stability, permeability, etc.[1] Deoxy-fluorination is an important method to convert alcohols to corresponding aliphatic fluorides. A variety of deoxyfluorination reagents have been developed, among which diethylamino-sulfur trifluoride (DAST) is the most used. But DAST has poor thermal stability with explosive potential. In recent years, safer deoxyfluorinating reagents have been developed, such as PyFluor[2], AlkylFluor[3] etc. Yet, the choice of base is also very important with deoxy-fluorination. For example, PyFluor and DBU are reported to be the best combination of acyclic secondary alcohol deoxyfluorination reaction, which can minimize the generation of by-products[2].
For MedChem synthesis, we need to quickly identify better choices of reagent and base for deoxyfluorination of the substrates of our interest. In 2018, Professor Abigail G. Doyle of Princeton University reported the development of a Quantum Mechanics (QM) and Machine Learning (ML) tool for reliable prediction of the optimal reaction conditions of untested substrates for deoxyfluorination[4a].
By now, we are familiar with QM analyses of reactions. But what is machine learning (ML)?
ML is a field of inquiry devoted to understanding and building methods that ‘learn’. Its learning algorithms build models based on sample data, known as training data, in order to make predictions without being explicitly programmed to do so.
Figure 1 illustrates the process of building the QM-ML model for deoxyfluorination discussed in this chapter[4a, 5]. First, relevant data were collected for 640 screening reactions, including experimental yields, characteristic data of alcohols, sulfonyl fluorides, and bases used in the reactions. This includes QM calculated molecular and atomic parameters of the alcohols, e.g., LUMO and HOMO energies, electrostatic charge, carbon NMR data, dipole moment, etc. and their structural classification, e.g., primary, secondary, tertiary, cyclic, benzylic, allylic, homobenzylic, homoallylic, etc. Then 70% of the data was used as a training set for machine learning with Random Forest algorithm. The resultant model was then evaluated with the remaining 30% of the data. The validated QM-ML model could be used for prediction of optimal conditions for deoxyfluorination for alcohol substrates that are not in the data set.
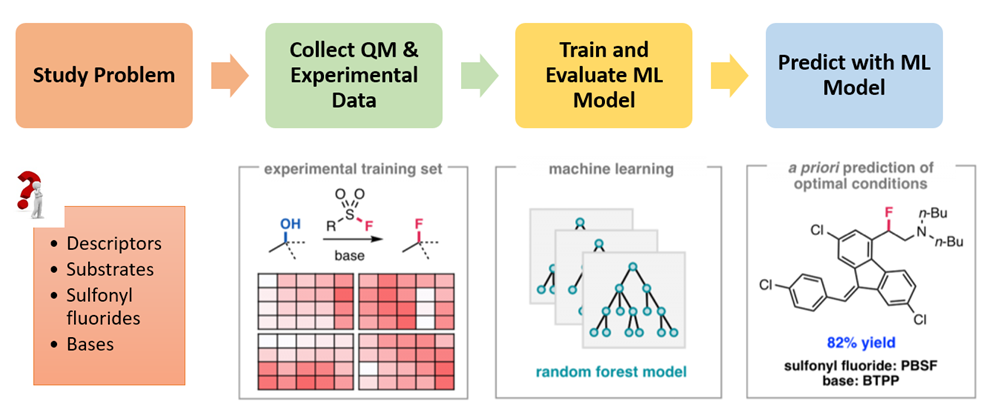
Figure 1. Process for building the QM-ML model for deoxyfluorination[4a, 5]
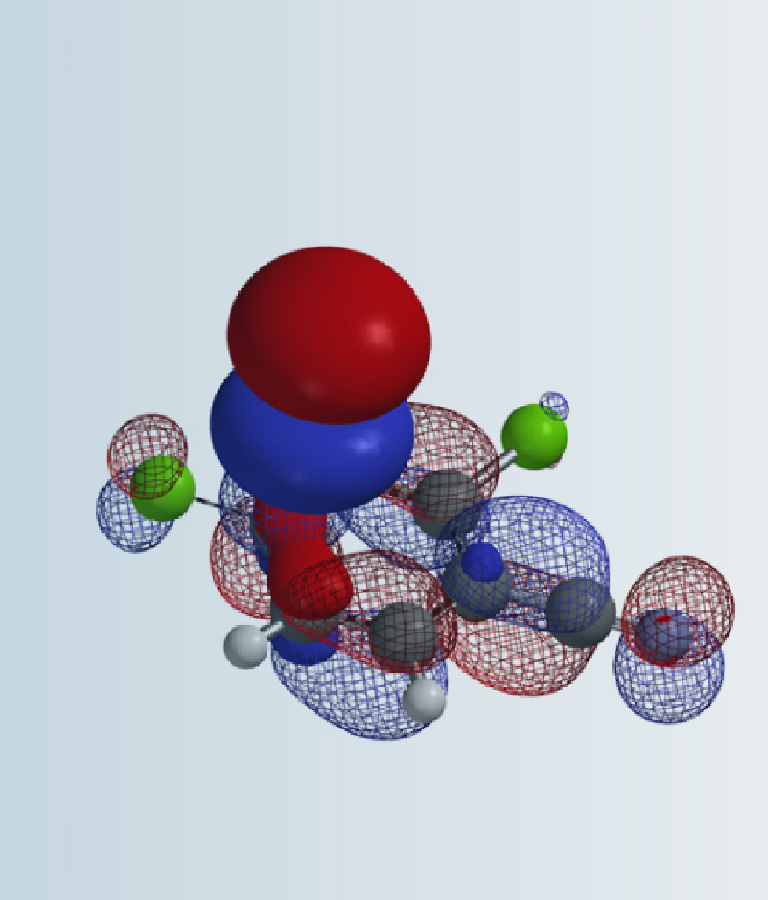
Figure 2 is the flow chart of using the QM-ML model for predicting reaction conditions of deoxyfluorination. The HOMO and LUMO energies of the alcohol reaction substrate, as well as the electrostatic charge and exposed area of the carbon attached to the hydroxyl group were first calculated with QM. It took a few minutes to create an input file with the above four parameters as well as the classification information of alcohols, and a few seconds for the QM-ML model to generate predicted yields for the 20 reaction conditions. A relatively simple process!
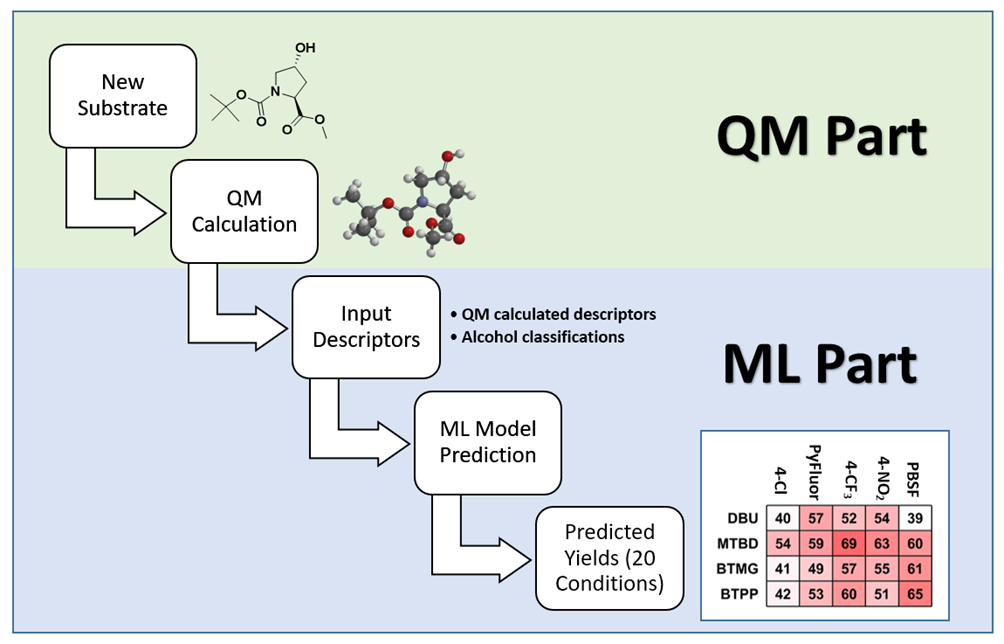
Figure 2. Workflow for using the QM-ML deoxyfluorination model for prediction[4b]
When the QM-ML paper was published, we were converting the precious alcohol 1 to the corresponding mesylate, and then using TBAF displacement to provide the desired fluoride 2, with an isolated yield of 43%, losing half of the optically pure alcohol 1.
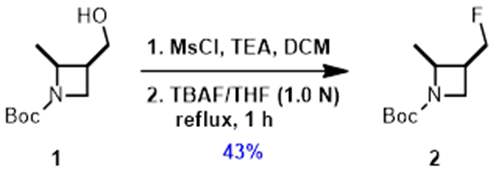
Figure 3. Two step procedure for deoxyfluorination of alcohol 1
Our prior knowledge in the use of QM for reaction analyses enabled us to quickly deploy Doyle’s deoxyfluorination model, which suggested the combination of perfluorobutansulfonyl fluoride (PBSF) and (tert-butylimino)tris(pyrrolidino)phosphorane (BTPP) as optimal conditions, with a predicted yield of 42.3%.
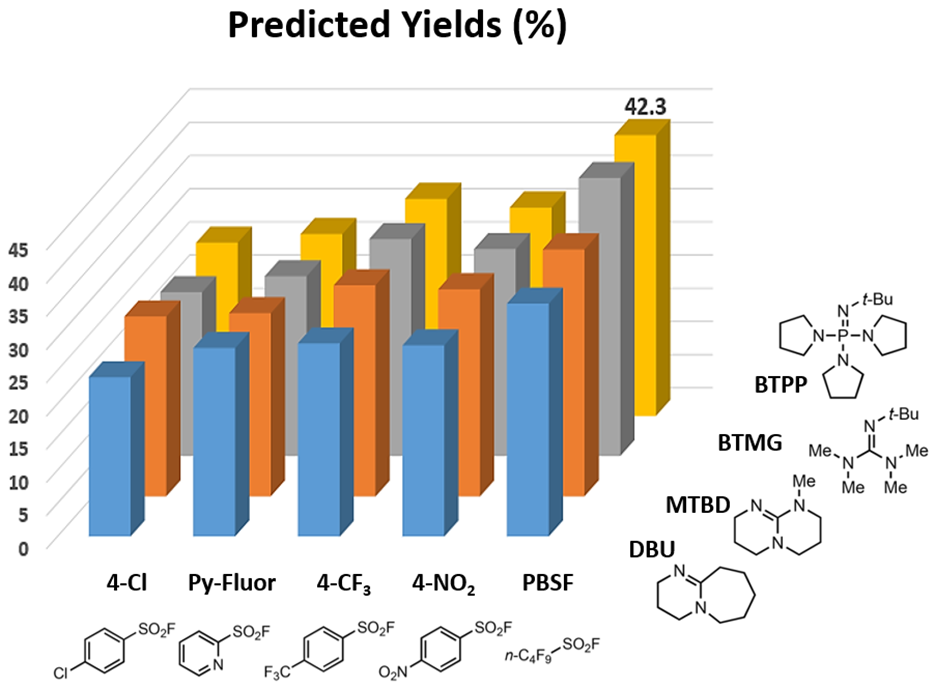
Figure 4. Output from QM-ML deoxyfluorination model for alcohol 1[4b]
Using the recommended PBSF and BTPP conditions, an isolated yield of 83% was achieved, nearly doubled in yield compared to the earlier two step procedure. Our first application of the QM-ML deoxyfluorination tool in our laboratory is quite successful.
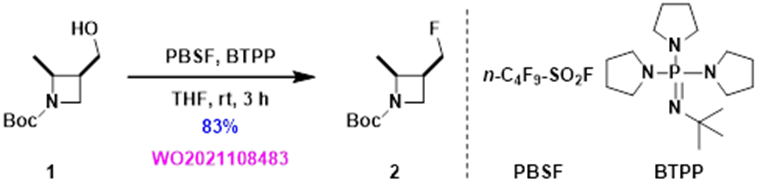
Figure 5. Deoxyfluorination result with suggested reaction conditions from QM-ML tool
With the QM-ML deoxyfluorination model, Doyle’s group found optimal reaction conditions for more than 30 alcohols, including active alcohols (benzylic alcohols, allylic alcohols), inactive alcohols, acyclic alcohols, cyclic alcohols, etc. However, for acyclic secondary alcohol 3, even though the optimal conditions predicted with QM-ML tool was the combination of 4-CF3PhSO2F and MTBD with a yield of 69%, experimentally the optimal reaction condition was found to be the combination of PyFluor and DBU[4,2].
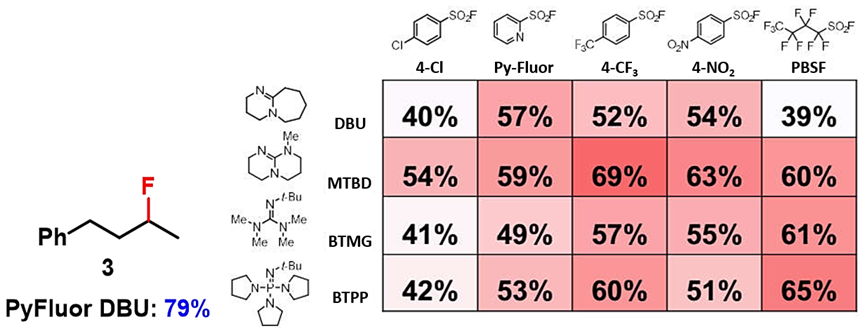
Figure 6. Deoxyfluorination of alcohol 3[4b]
In summary, combination of QM and ML has generated a very useful model for accurate prediction of optimal conditions for deoxyfluorination and isolated yields. We look forward to development of similar QM-ML reaction conditions predictive tools for other reactions.
We offer the above Deoxyfluorination Prediction as service (Figure 7). If your chemistry requires such analysis, please contact us by email at IDSU_Operation@wuxiapptec.com
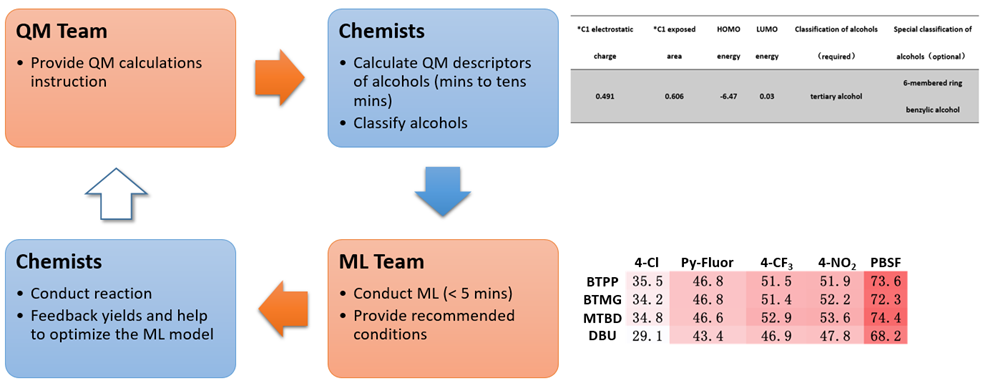
Figure 7. Workflow of our Deoxyfluorination Prediction Service
In general, the predicted yields from the QM-ML model have excellent correlation with experimental results (Figure 8). There are some notable exceptions.
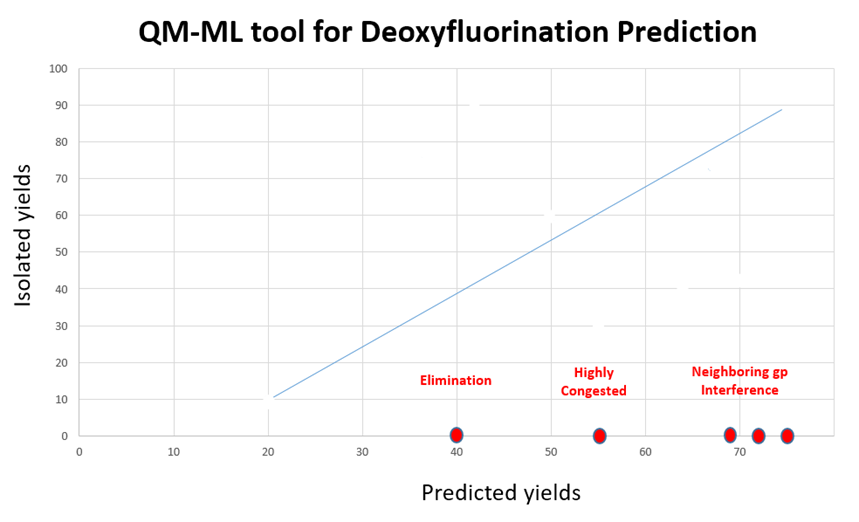
Figure 8. Comparison of QM-ML predicted and isolated yields with our deoxyfluorination
For highly active and easily eliminating alcohols and tertiary alcohols, the above QM-ML model does not provide useful deoxyfluorination conditions. Moreover, we found Sulfox-Fluor[6] works well for alcohols with high activity and easy elimination; Fluolead[7] and XtalFluor-E[8] are useful for tertiary alcohols. If you are aware of other excellent reagents, please share them with us.
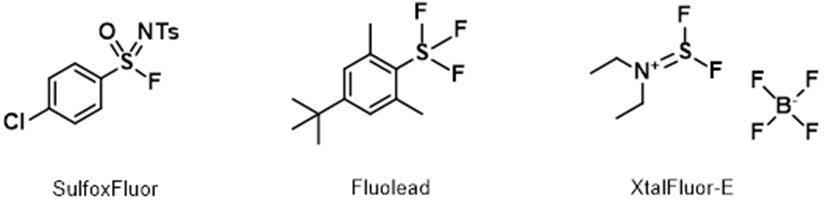
Figure 9. Other useful deoxyfluorination reagents
References:
[1] D.E. Yerien, S. Bonesi, A. Postigo, Org. Biomol. Chem. 2016, 14, 8398.
[2] M.K. Nielsen, C.R. Ugaz, W. Li, A.G. Doyle, J. Am. Chem. Soc. 2015, 137, 9571.
[3] N.W. Goldberg, X. Shen, J. Li, T. Ritter, Org. Lett. 2016, 18, 6102.
[4] a. M.K. Nielsen, D.T. Ahneman, O. Riera, A.G. Doyle, J. Am. Chem. Soc. 2018, 140, 5004. b. The three substituted (Cl, CF3, and NO2) phenylsulfonyl fluorides are para substituted instead of meta substituted as described on p 5004 of this paper. They were prepared from commercially available para sulfonyl chloride. See paper’s supporting information S-21.
[5] F. Strieth-Kalthoff, F. Sandfort, M.H.S. Segler, F. Glorius, Chem. Soc. Rev. 2020, 49, 6154.
[6] J.K. Guo, C.W. Kuang, J. Rong, L.C. Li, C.F. Ni, J.B. Hu, Chem. Eur. J. 2019, 25, 7259
[7] T. Umemoto, R.P. Singh, Y. Xu, N. Saito. J. Am. Chem. Soc. 2010, 132, 18199.
[8] S. Kalidindi, A.S. Gangu, S. Kuppusamy, S. Sathasivam, V. Shekarappa, S. Murugan, S. Bondigela, M. Kandasamy, K. Ghanta, A. Vinodini, A. Shrikant, R. Ramachandran, W.P. Gallagher, N. Kopp, F. González-Bobes, M.D. Eastgate, R. Vaidyanathan, Org. Process Res. Dev. 2021, 25, 1556.