 Choose language
Choose language
< Return to main menu
Choose language



News & Events

Services & Solutions
News & Events
Career
Platform
Magical Power of Quantum Mechanics
Events
News
Magical Power of Quantum Mechanics
Divergence in haloselectivity of polyhalogenated substrates is a long-standing puzzle in organic chemistry. For example, nucleophilic aromatic substitutions of 2,5-dibromopyridine (see Chapters 1, 7, 10, and 19 for analyses of observed selectivity) and palladium-catalyzed cross coupling reactions occur preferentially at C-2 [1,2] (see Chapter 5 for analysis), while halogen-metal exchange reactions occur preferentially at C-5 (Figure 1)[3,4].
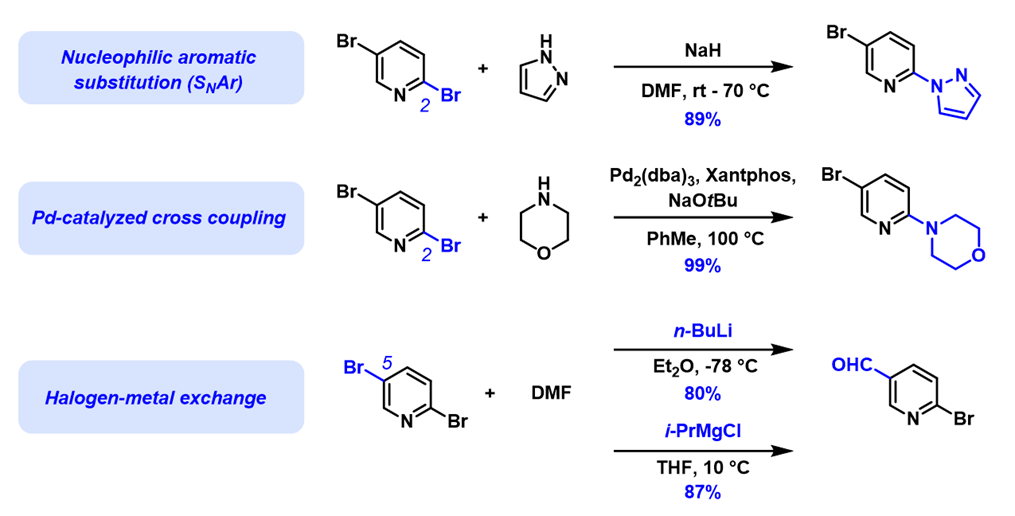
Figure 1. Haloselectivity: SNAr and cross coupling vs halogen-metal exchange reactions of 2,5-dibromopyridine.
How to account for this and other similar divergent examples reported in the literature[5,6]? What is the quantum origin of these divergences in reactivity?
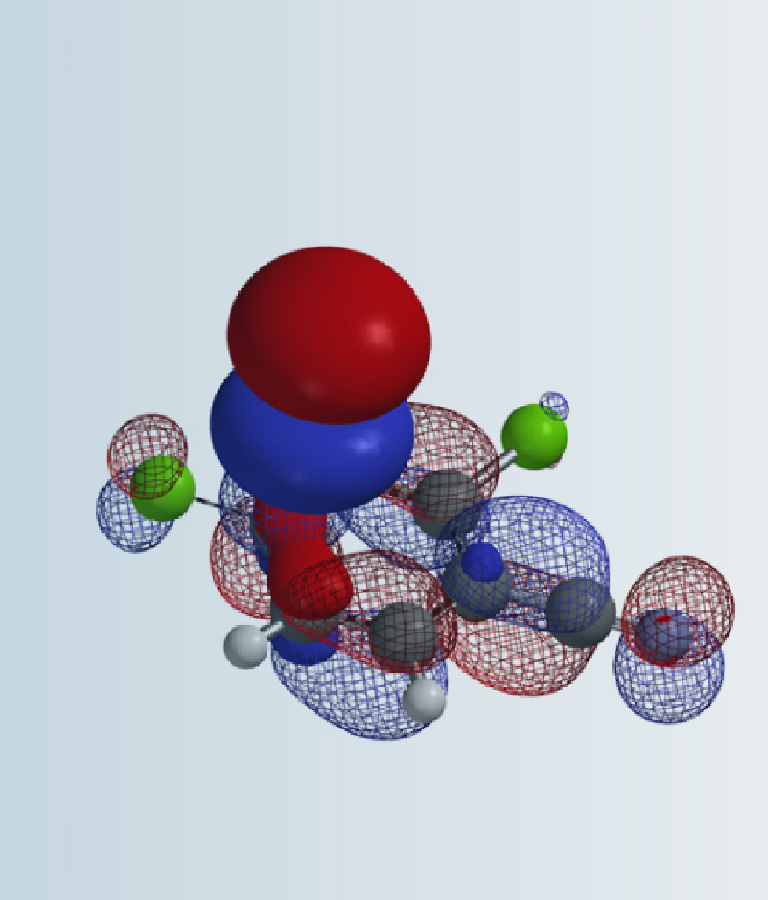
Let’s start by looking at the LUMO of bromoethane (Figure 2). Its lobe on the opposite side of the C-Br σ-bond interacts with HOMO of incoming nucleophile for SN2 reaction. The small lobe on the C-H bond at the C2 enables interaction with base for elimination reaction. Then the distinctive four LUMO lobes along the C-Br bond, with the outer lobe extending further in space, resemble a String-of-Pearls. We reasoned that organic halides use unoccupied orbitals with this characteristic feature for halogen-metal exchange reactions and metalation, and examined orbitals with such feature to account for/predict the distinctive haloselectivity observed in halogen-metal exchange reactions.
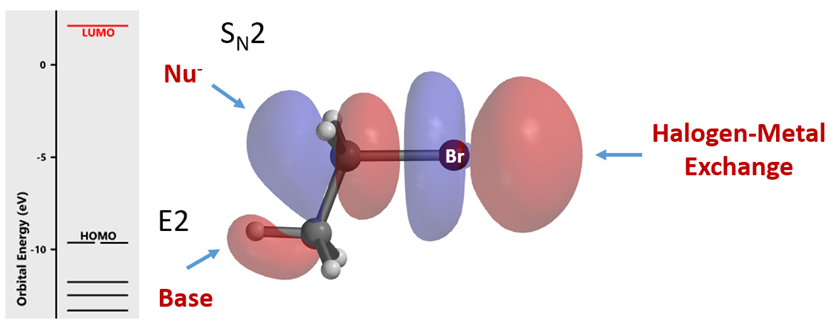
Figure 2. LUMO of bromoethane and chemical reactions associated with its specific lobes[7]
First, let’s see whether we can account for the selectivity observed with the bromine-metal exchange reaction of 2,5-dibromopyridine. Its LUMO, LUMO+1, and LUMO+2 are shown in Figure 3. The “String-of-Pearls” shaped lobe distribution along the C-Br bond is only observed with LUMO+2, not with LUMO nor LUMO+1. The LUMO+2 terminal lobe on C5-Br bond is large and protrudes beyond its Electron Density isosurface (0.002 e/au3; 99.45%). On the other hand, such a distinctive pattern is not obvious along the C2-Br bond. As such, we reasoned that C5 bromide can preferentially interact with metal reagents, accounting for the C5 haloselectively observed with the bromine-metal exchange reaction of 2,5-dibromopyridine.
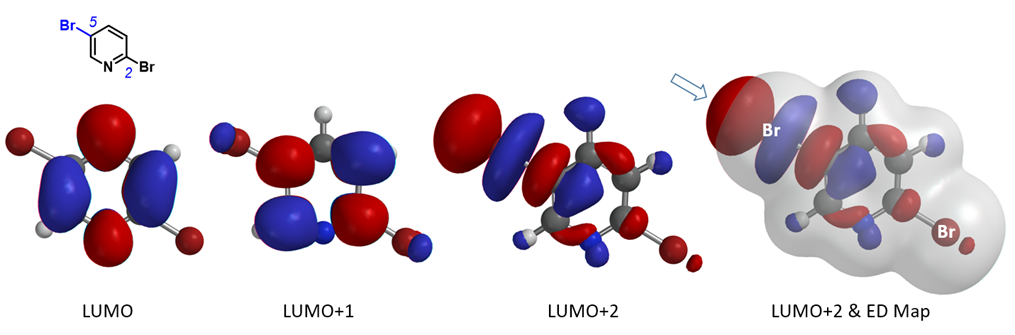
Figure 3. 2,5-Dibromopyridine: LUMO, LUMO+1, LUMO+2, and overlay of LUMO+2 with Electron Density Map[7]
Next, let’s apply the analysis to additional examples (Figure 4). With 1,2,4-tribromobenzene, its LUMO+2 has three Strings-of-Pearls, with the largest set along the C2-Br bond, and its terminal lobe extends beyond the isosurface, consistent with C2 selective metalation observed. Similarly for 2,3-dibromothiophene, its LUMO+1 lobes along the C2-Br bond are larger than those on the C3-Br bond, accounting for its selective C2 functionalization. For N-methyl-3,5-dibromo-pyrazole and N-methyl-5,7-dibromoindole, the characteristic LUMO lobes are significantly larger at C5 vs C3 bromide and C7 vs C5 bromide, respectively, aligned with the haloselectivity observed. These examples also exemplify a unique characteristic of halogen-metal exchange reactions, that is, due to the unique unoccupied orbitals involved, with their terminal lobes extending beyond Electron Density isosurfaces, they are not sensitive to neighboring group’s steric hindrance.
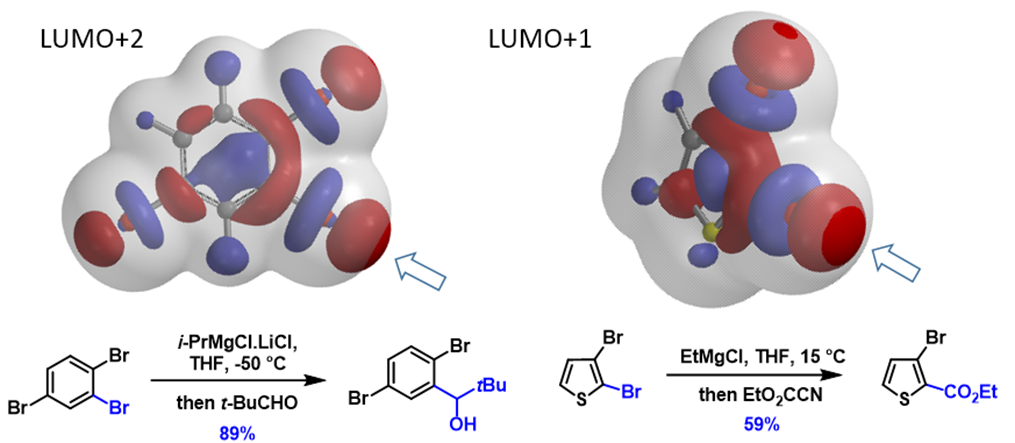
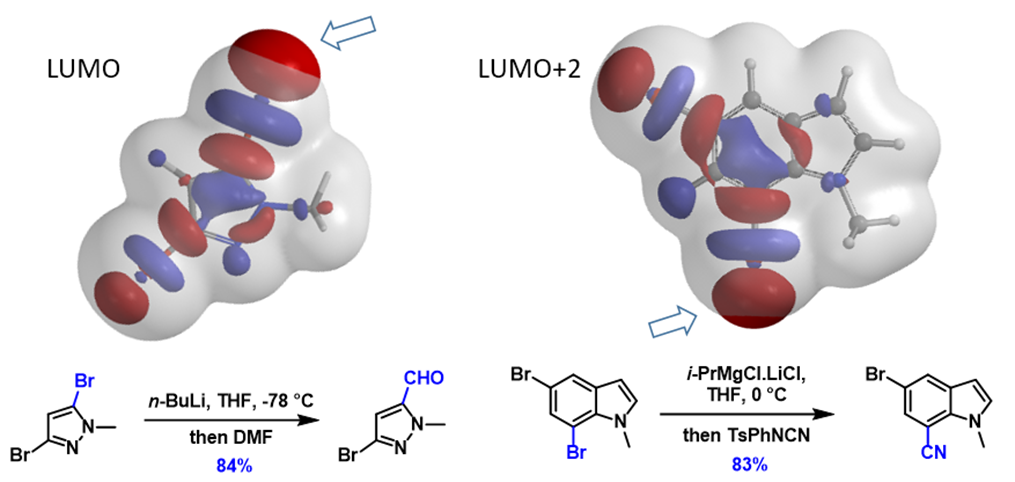
Figure 4. “Pearl-of-String” shaped LUMO or LUMO+n lobes for predicting halogen-metal exchange reactions[7a-d]
This method can also be extended to direct metalation reactions (Figure 5). With 2,4-dichloro-5-fluoropyrimidine, there are two chloro groups. The terminal lobe on the C4-Cl bond extends beyond the isosurface of the molecule while the lobe along the C2-Cl bond does not. This enables selective formation of the corresponding C4-Zn reagent, which reacts with acetic acid, and leads to the selective dechlorination observed.
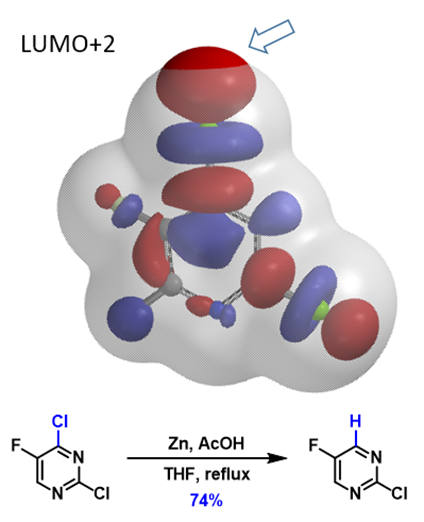
Figure 5. Dechlorination of 2,4-dichloro-5-fluoropyrimidine (LUMO+2 and Overlay with Electron Density Map)[7e]
This LUMO analysis is further validated with >100 examples (reaction solvent THF or Et2O) reported in the literature, enabling us to predict with high accuracy the haloselectivity in halogen-metal exchange reactions.
With LUMO+2 and LUMO+3 usually at significantly higher energy levels than LUMO and LUMO+1, one may wonder how they can be involved in these reactions. For feasibility of reactions, the key determining factor is the HOMO-LUMO energy gap of the reactants, not their absolute numbers (see QM Chapter 25)[8]. Lithiation and Grignard reagents have high HOMO energy, the resulting energy gaps are within the range where these reactions will proceed. Lithiation reagents usually have higher HOMO energy than Grignard reagents, consistent with the observations that lithium-halide exchange will proceed at lower temperature than the Grignard exchange reactions.
When the characteristic LUMO or LUMO+n lobes are similar in size and accessibility, we found that relative energies calculated for regioisomers of corresponding carbanions correlate well with the selectivity observed. The regioisomer with lower carbanion energy will have a higher proportion in the product mixture[9]. This is a convenient approximation for prediction. The halogen-metal exchange product should exist with carbon-metal bonds, and their stabilities are affected by various factors such as solvent, temperature, and directing groups.
This can be exemplified with 2,3-dibromobenzothiophene (Figure 6) with two sets of similarly sized LUMO+1 String-of Pearls. Calculated relative energies of C2 vs C3 carbanion are 0.00 vs 11.66 kcal/mol, respectively, consistent with the observed selective C2 functionalization[10].
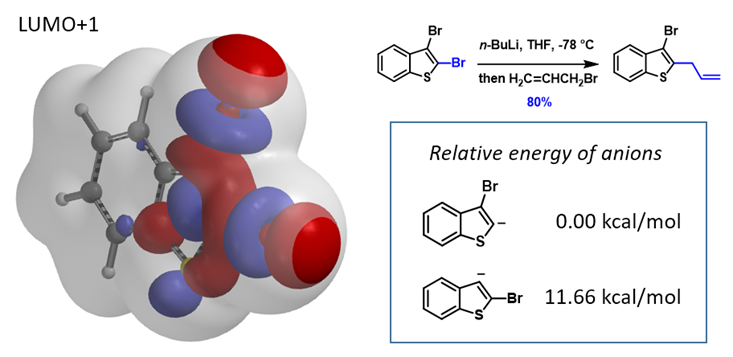
Figure 6. Prediction of haloselectivity of 2,3-dibromobenzothiophene with relative energy of carbanion[10]
In summary, Halogen-Metal Exchange reactions involve unique “String-of-Pearls” shaped LUMO lobes along carbon-halogen bonds of the substrates. Comparison of the size of these unique orbitals/lobes enables us to predict the regioselectivity of the exchange. These unique lobes may be on LUMO, LUMO+1, LUMO+2, or LUMO+3. When the LUMO lobes are similar in size, the relative energy of the corresponding carbanion can be used for differentiation. The differences in haloselectivity of Oxidative Addition and SNAr vs Halogen-Metal Exchange arise from the differences in quantum origin of these reactions, i.e. the former uses LUMO lobe centered over the carbon of the C-X bonds, while the latter utilizes the “String-of-Pearls” large terminal lobes that protrude beyond the Electron Density isosurface.
Expanding our analyses to include LUMO+n enabled us to appreciate why haloselectivity of Halogen-Metal Exchange could be so different. A decades-old chemistry puzzle is solved.
Shown in Figure 7 is ethyl 2,5-dibromothiazole-4-carboxylate with its two low energy conformations. LUMO+1 “String-of-Pearls” analyses of them suggested opposite halo-selectivity. Experimentally, the bromine-Grignard exchange is C5 selective[11]. What could be the controlling factor for this transformation?
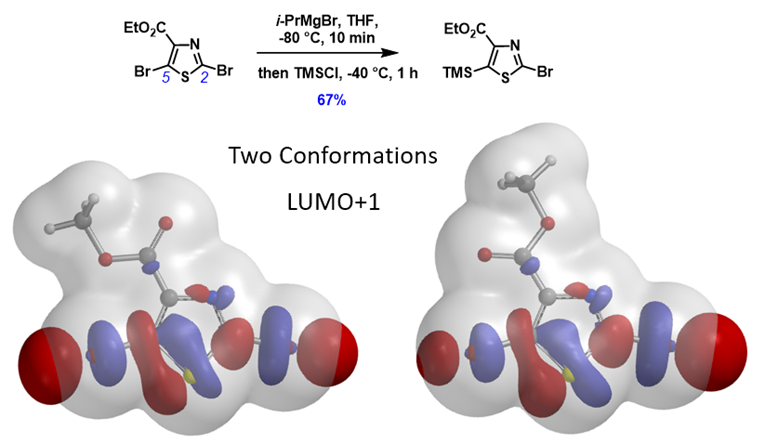
Figure 7. Two conformations of ethyl 2,5-dibromothiazole-4-carboxylate: LUMO+1 and ED map overlay[11]
References:
[1] C.S. Rani, A.G. Reddy, E. Susithra, K.K. Mak, M.R. Pichika, S. Reddymasu, M.V.B Rao. Med. Chem. Res., 2021, 30, 74.
[2] J.G. Ji, T. Li, W.H. Bunnelle, Org. Lett., 2003, 5, 4611.
[3] P.N.W. Baxter. Chem. Eur. J., 2003, 9, 5011.
[4] L. Peng, J.D. Wen, C.G. Pan, X.W. Pan, Y. Xie, W. He, Q.F. Lin, X.Y. Zhang (Chongqing Sansheng Industrial Co., Ltd., China), CN 112479991 A, 2021.
[5] J.A. Joule & K. Mills, Heterocyclic Chemistry 4th Ed. Malden, MA, USA: Blackwell Publishing Ltd., 2000; pp 26-45.
[6] https://www.scripps.edu/baran/images/grpmtgpdf/Gutekunst_Apr_10.pdf
Divergences in haloselectivity highlighted with 3,6-dibromoindole (pp 2), 2,3,5-tribromo thiophene (pp 3), 2,5-dibromopyridine (pp 6), in palladium catalyzed cross coupling reactions vs halide metal exchange reactions.
[7] LUMO, LUMO+1, LUMO+2 and Electron Density Map calculated with Spartan’20 DFT-ωB97X, 6-31G*. (a) A. Krasovskiy, P. Knochel, Angew. Chem. Int. Ed., 2004, 43, 3333. (b) C. Christophersen, M. Begtrup, S. Ebdrup, H. Petersen, P. Vedsø. J. Org. Chem., 2003, 68, 9513. (c) Y. Yin, C.J. Chen, R.N. Yu, L. Shu, T.T. Zhang, D.Y. Zhang, Bioorg. Med. Chem., 2019, 27, 1562. (d) P. Anbarasan, H. Neumann, M. Beller, Chem. Eur. J. 2011, 17, 4217. (e) C. De Savi, A. Pape, J.G. Cumming, A. Ting, P.D. Smith, J.N. Burrows, M. Mills, C. Davies, S. Lamont, D. Milne, C. Cook, P. Moore, Y. Sawyer, S. Gerhardt, Bioorganic Med. Chem. Lett., 2011, 21, 1376.
[8] L.G. Zhuo, W. Liao, Z.X. Yu, Asian J. Org. Chem. 2012, 1, 336.
[9] (a) H.J.S. Winkler, H. Winkler, J. Am. Chem. Soc., 1966, 88, 964. (b) H.J.S. Winkler, H. Winkler, J. Am. Chem. Soc., 1966, 88, 969. (c) H.R. Rogers, J. Houk, J. Am. Chem. Soc., 1982, 104, 522. (d) K.B. Kenneth, S. Sklenak, F.B. William, J. Org. Chem., 2000, 65, 2014.
[10] Relative energy of anion calculated with Spartan’20 DFT-ωB97X, 6-31G*, nonpolar solvent. T.P. Sura, D.W.H. MacDowell, J. Org. Chem., 1993, 58, 4360.
[11] M. Abarbi, J. Thibonnet, L. Bérillon, F. Dehmel, M. Rottländer, P. Knochel, J. Org. Chem., 2000, 65, 4618.
This article is written and edited by Dong Pan, Jun Liu, Wenfeng Liu, Tommy Lai, Yongsheng Chen, John S. Wai.