 Choose language
Choose language
< Return to main menu
Choose language



News & Events

Services & Solutions
News & Events
Career
Platform
Magical Power of Quantum Mechanics
Events
News
Magical Power of Quantum Mechanics
Bioconjugation of proteins with thio modification of cysteine residues usually exhibits excellent chemoselectivity as free cysteines rarely occur on protein surface. Electrophiles, such as iodoacetamide, maleimide, and disulfide derivatives are usually used. Shown on Figure 1 is an excellent example in which the iodoacetamide and maleimide strategies are being used sequentially on XTEN to achieve high Drug-to-antibody ratio (DAR) [1]. A recent report showed that chloroacetamide exhibits even greater specificity than iodoacetamide for cysteine residues in bioconjugation[2]. In this chapter we’ll discuss the difference observed in reactivity between N,N-dimethyl and N-methyl chloroacetamide in thiol conjugation and results from our QM analyses.
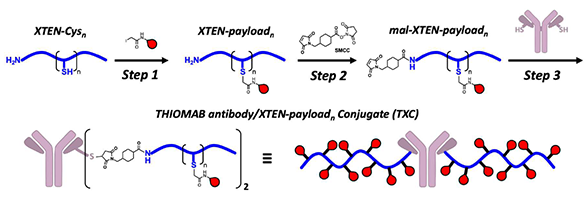
Figure 1. A thiol bioconjugation strategy for high Drug-to-antibody ratio (DAR).
Shown in Figure 2 is the difference in reactivity observed between N,N-dimethyl and N-methyl chloroacetamides with a thiol. Intuitively, we were expecting the opposite, that is, the incoming thio will face more steric hindrance with the tertiary amide. To understand why, we first examined LUMO and LUMO map of the two acetamides (Figure 3).
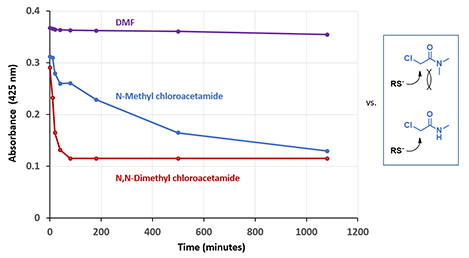
Figure 2. Difference in reactivity observed between N,N-dimethyl and N-methyl chloroacetamides with a thiol vs expectation.
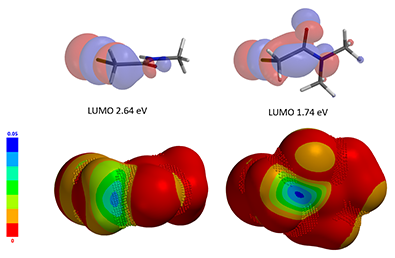
Figure 3. LUMO and LUMO map of N,N-dimethyl and N-methyl chloroacetamides (IsoVal 0.002 e/au3, 99.2%).
LUMO energy of N-methyl chloroacetamide is 2.64 eV. Its LUMO map and inaccessibility marker suggest that access to the LUMO lobe across on the C-Cl carbon is limited. On the other hand, LUMO energy of N,N-dimethyl chloroacetamide is lower, 1.74 eV, and the LUMO lobe across on the C-Cl carbon is more exposed and accessible, suggesting that it shall be more reactive, consistent with experimental observations. Done? We reasoned that there should be additional reasons and decided to conduct further QM analyses of this pair of reactions.
First, we modeled for the reaction energy profile of N,N-dimethyl chloroacetamide, using methyl thiolate as cysteine surrogate to speed up our calculations (DFT B3LYP-D3/6+31G*, the same below).
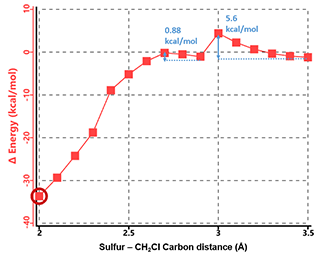
Figure 4. Reaction energy profile of N,N-dimethyl chloroacetamide and methyl thiolate.
As shown in Figure 4, there are two relatively low energy barriers, 5.60 and 0.88 kcal/mol, associated with the reaction, peaking at sulfur to CH2Cl carbon distance of 3.0 Å and 2.6 Å, respectively, while N,N-dimethyl chloroacetamide undergoes remarkable conformational changes. When the sulfur and carbon are 3.5 Å apart, the carbon-chlorine bond is almost coplanar with the amide group. Thio displacement of the chloride requires the nucleophile to approach from the backside, and faces steric hindrance from the amide methyl group as energy peaks at 3.0 Å. The barrier is overcome at 2.9 Å, when the carbon-chlorine bond rotates perpendicular to the amide plane and lines up perfectly for the nucleophilic substitution reaction, with a second energy barrier of only 0.88 kcal/mol.
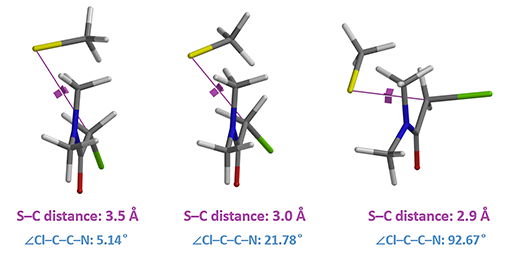
Figure 5. Conformational and dihedral angle Cl–C–C–N changes in reaction with N,N-dimethyl chloroacetamide.
Similarly, we calculated for the reaction energy profile of N-methyl chloroacetamide (Figure 6), which exhibits a single energy peak. At ~ 10 kcal/mol, it is significantly higher than that of N,N-dimethyl chloroacetamide. Both chloroacetamides undergo the same remarkable conformational changes in the process, that is, the carbon-chlorine bond rotates from almost coplanar to perpendicular to the amide group (Figure 7, top).
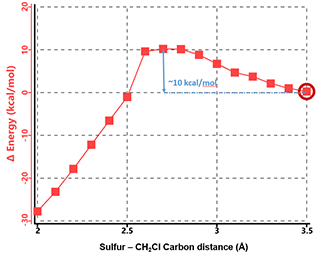
Figure 6. Reaction energy profile of N-methyl chloroacetamide and methyl thiolate.
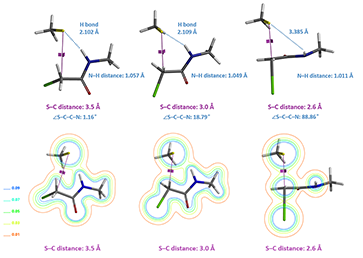
Figure 7. Conformational, dihedral angle Cl–C–C–N, and electron density changes in reaction with N-methyl chloroacetamide.
However, for N-methyl chloroacetamide, the incoming thiolate hydrogen bonds with the amide NH (Figure 7, S-C distance 3.5 and 3.0 Å), as indicated by the S···H–N bond distances and electron density slices calculated. For the nucleophilic substitution to proceed further, this hydrogen bond needs to be broken, incurring energy cost to reach transition state (Figure 7, S-C distance 2.6 Å), consistent with lower reactivity observed with N-methyl chloroacetamide.
We learned from the example in chapter 26 that hydrogen bond interactions could stabilize transition state, accelerating the rate of the reaction. In this chapter, we learned that when the hydrogen bond stabilizes the reacting complex at early stages and needs to be broken to reach transition state, it could slow down the reaction. For chloroacetamide bioconjugation on cysteine residues, this turns out to be a good thing.
For organic chemistry, we first consider the steric and electronic effect of reactants and reagents at ground state, then intuitively evaluate potential non-covalent interactions that could have a significant impact on the reaction process. QM enables us to quantify such interactions to account for our observations, improves our intuition, and guides our chemistry.
SNAr reactions usually proceed more readily in polar aprotic solvents (DMSO, MeCN, DMF, etc) than in polar protic ones, as shown for SNAr reactions of 1-fluoro-4-nitrobenzene with azide (Table 1)[4].
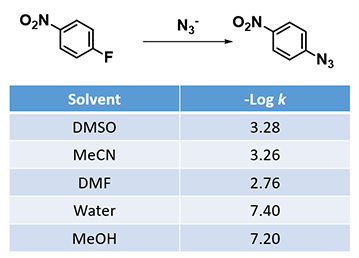
Table 1: Reaction rate constants (log k) of n-Bu4N3 with 1-fluoro-4-nitrobenzene in different solvents at 25 ºC[4]
In our laboratories, we encountered remarkable SNAr reactions that did not proceed at all in DMSO, NMP, MeCN, but proceeded readily in alcohols, as exemplified by the reaction of primary amines with 2-fluoropyridine. Since primary amines are highly solvated in polar protic solvents, their reactions in alcohol were anticipated to be slower than in polar aprotic ones. Why are we observing the opposite?
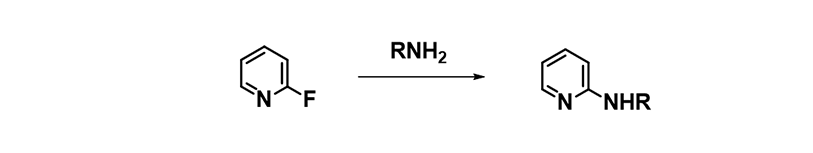
Figure 8. SNAr Reaction of 2-fluoropyridine with primary amines
This article is written and edited by Shouliang Wang, Zhong Zheng, Qiuyue Wang, Jian Wang, Yongsheng Chen, John S. Wai
References:
[1] https://www.researchsquare.com/article/rs-356231/v1
[2] J. Kalia, R. T. Raines, Curr. Org. Chem. 2010, 14, 138.
[3] P. Atkins, J. de Paula, J, Keeler, Atkins’ Physical Chemistry (11th ed.). Oxford University Press. Oxford, 2018: pp 584
[4] B.G. Cox, A.J. Parker, J. Am. Chem. Soc. 1973, 95, 408.