 Choose language
Choose language
< Return to main menu
Choose language



News & Events

Services & Solutions
News & Events
Career
Platform
Magical Power of Quantum Mechanics
Events
News
Magical Power of Quantum Mechanics
1,2,3-Triazole is a common heterocycle in medicinal chemistry, providing us novel structures with a wide range of biological activities. 1,4-Disubstituted 1,2,3-triazoles are versatile (Figure 1): they have been used as part of the pharmacophores; as bioequivalent for specific functional groups; as peptidomimetic to improve metabolic stability against proteases; and as linkers for multispecific drugs, such as PROTAC®, drug conjugates, and so on [1].
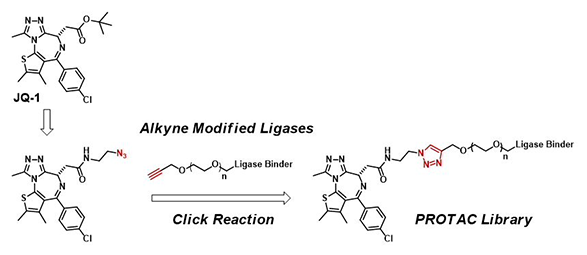
Click-Chemistry-mediated rapid synthesis of bispecific molecules for inducing protein degradation.
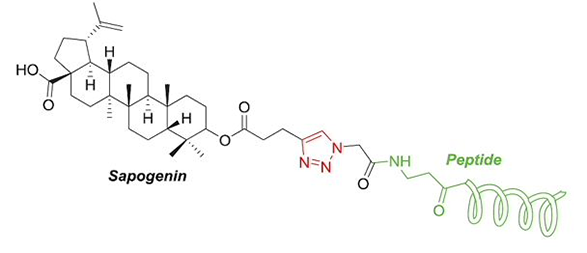
Click-Chemistry-mediated conjugation of a nonspecific antiviral Sapogenin with a HIV fusion inhibitor.
Figure 1. Examples of the use of 1,2,3-triazole moiety in drug design
1,2,3-triazoles are usually constructed with [3 + 2] cycloaddition reaction, including the Cu(I) catalyzed Click Reaction (Figure 2) developed by Professor Sharpless. The required azido compounds are highly reactive, yet usually associated with toxic and explosive properties. As such, safer methods for their generation and handling are highly desirable in MedChem discovery efforts.
Figure 2. Example of 1, 2, 3-triazole skeleton construction
In 2019, Jiajia Dong of Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, and Barry Sharpless published in Nature the use of fluorosulfuryl azide as a safer and more efficient reagent for the preparation of various azides and their use in the Click reaction (aka Double Click) to construct a triazole library with more than a thousand compounds (Figure 3).[2]
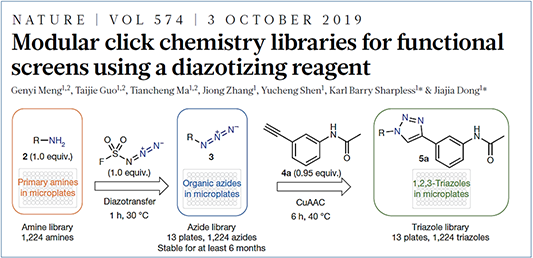
Figure 3. Diazo transfer reaction published in Nature, 2019
Fluorosulfuryl azide, FSO2N3, introduced in this paper shows superior efficiency in diazo transfer to primary amines. Compared with the traditional diazo transfer reagents, it presents many advantages: the reactions are carried out at room temperature, 1:1 stoichiometric, fast and efficient conversion, suitable for all primary amines, including tertiary alkyl amines (e.g. t-BuNH2), near quantitative conversion. In this chapter, we’ll discuss QM analysis of the reaction mechanism of this useful diazo transfer reagent.
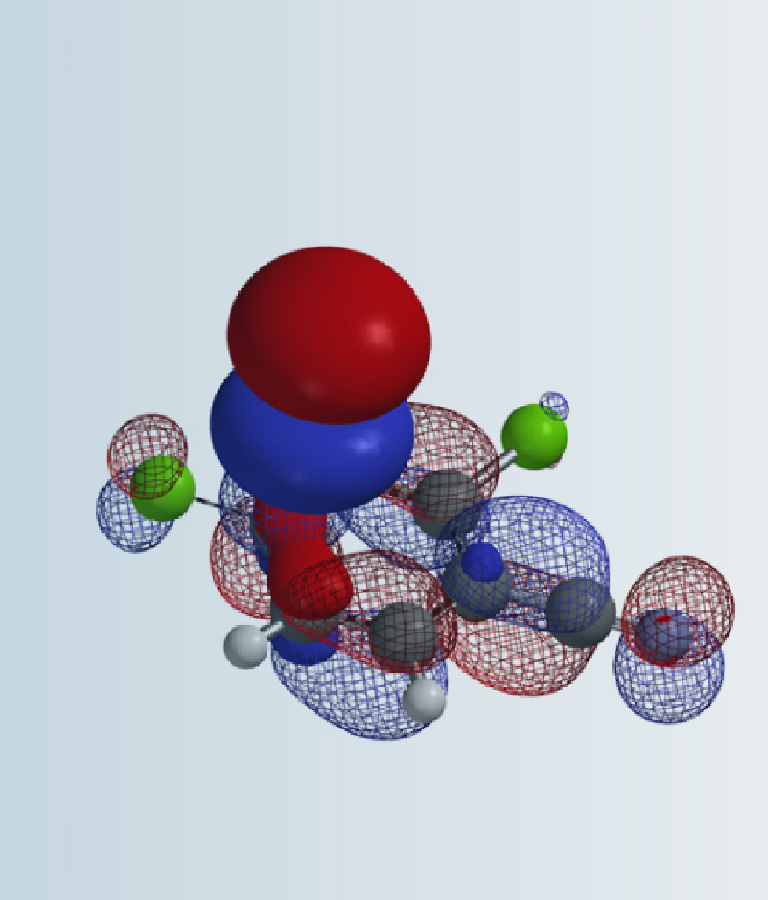
Fluorosulfuryl azide is highly electrophilic. Among the three azido nitrogen, we need to determine which nitrogen atom is more electrophilic to interact with the incoming amine (Fig. 4).
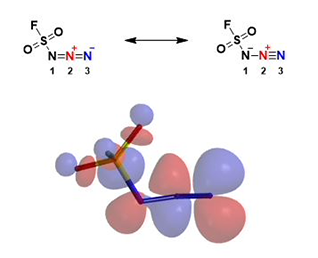
Figure 4. Resonance structures of fluorosulfuryl azide and its LUMO
The LUMO and LUMO map of fluorosulfuryl azide were calculated first (see Chapter 1). Results are shown on Figure 5, with LUMO lobe on N-3 > N-2 > N-1, and LUMO map clearly shows that N-3 is more accessible than N-2, suggesting that nucleophilic attack is more likely to occur on N-3. This is consistent with results from a 15N isotope labeling experiment reported by Samuelson et al. in 2014[3].
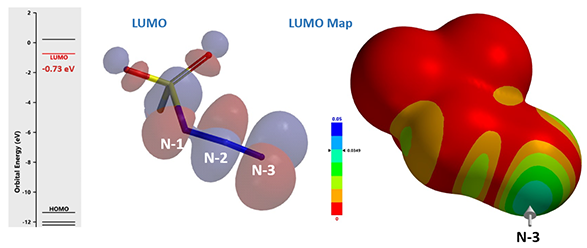
Figure 5. LUMO and LUMO map of fluorosulfuryl azide
The Nature paper pointed out that water is crucial for fluorosulfuryl azide to transfer a diazo group to amines efficiently. Our preliminary calculations indicated that nucleophilic attack of amine groups is a stepwise mechanism, not a one-step concerted mechanism (Figure 6), and shall include a water molecule in the reaction modeling, i.e. the amine attacks N-3 of fluorosulfuryl azide first, followed by transfer of a hydrogen atom from the amino group via a water molecule to N-1 of the reagent.
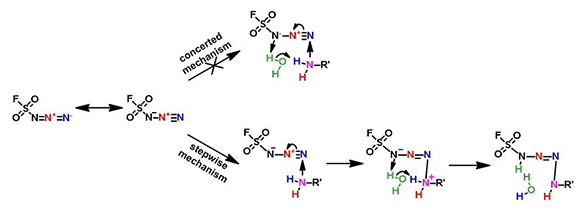
Figure 6. Possible mechanism for nucleophilic attack of amine on FSO2N3.
Shown on Figure 7-1 is the reaction energy profile calculated for the addition step, with the distance of the amine N—N-3 bond changing from 2.4 Å to 1.5 Å over 10 steps, and an energy barrier less than 5 kcal/mol. Next, we used the final structure in the addition step as the starting point to calculate for its reaction energy profile of the hydrogen atom transfer step. The results show that the activation energy of this step is about 15 kcal/mol, an energy barrier that could readily be overcome under room temperature conditions (Figure 7-2) [4]. The hydrogen transfer is relayed via a water molecule, with a seven-membered ring transition state.
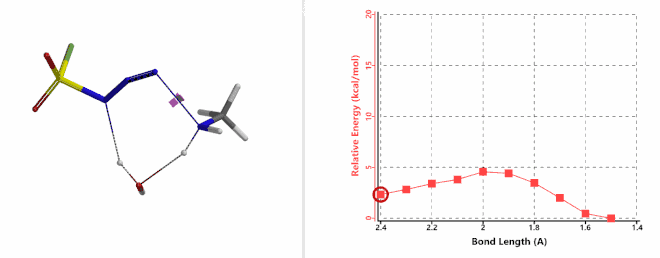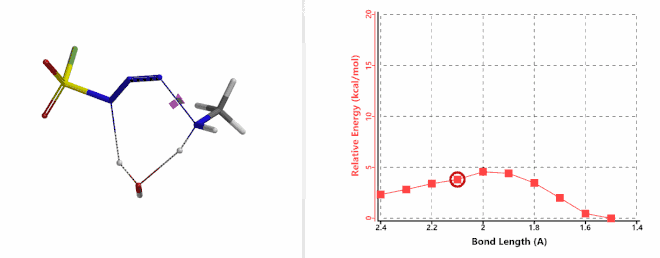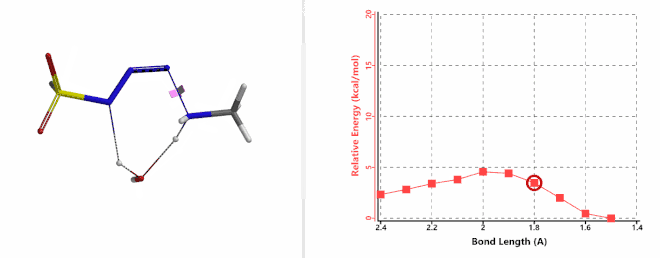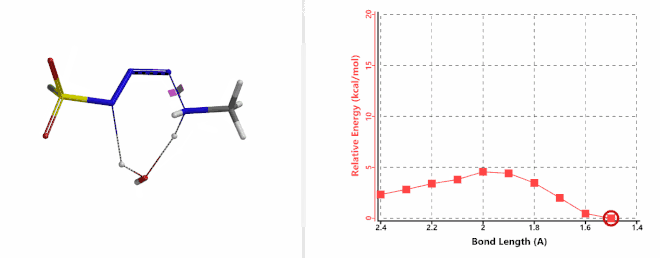
Figure 7-1. Reaction energy profile of amine addition to FSO2N3
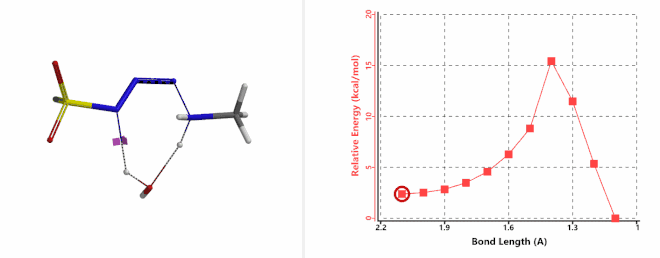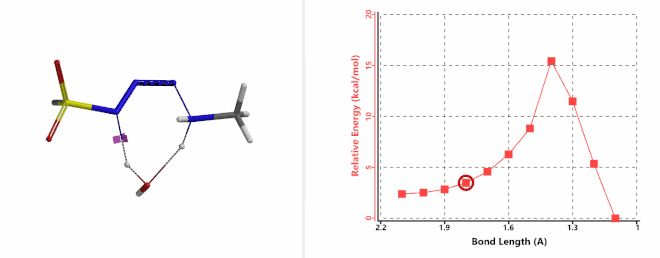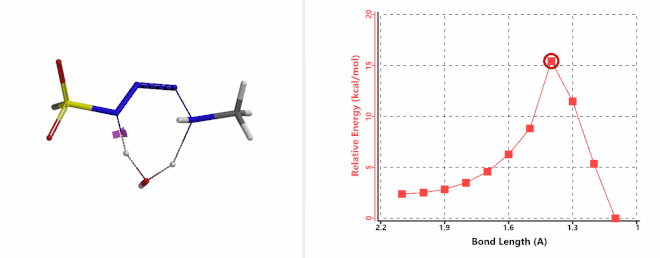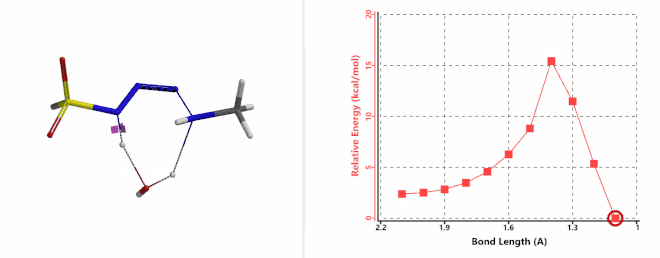
Figure 7-2. Reaction energy profile of hydrogen transfer via a water molecule
We selected the structures at the highest energy point from the two reaction energy profiles for more accurate Transition State and imaginary frequency calculations[5]. Resulting transition state structures are shown on Figure 8, with their respective imaginary frequency of 136 and 1280 cm-1 that are corresponding to the bonds being made and broken, satisfying the criteria for them to be considered as transition states.
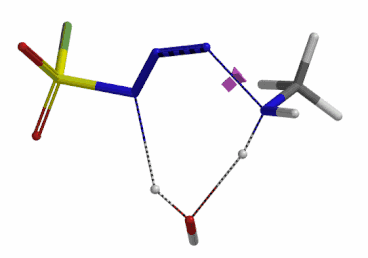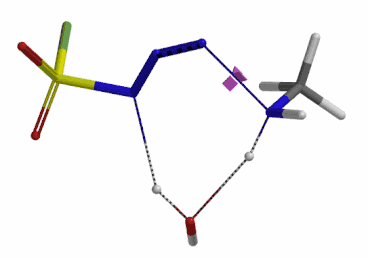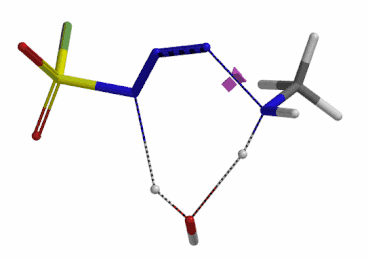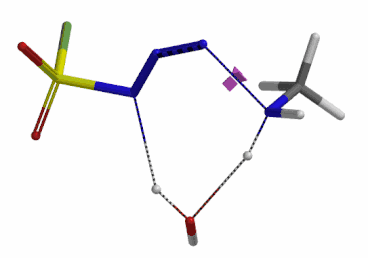
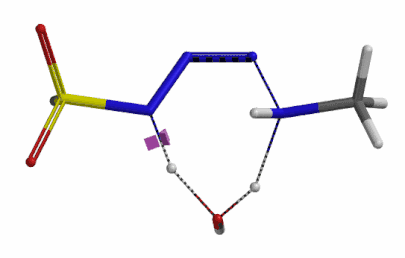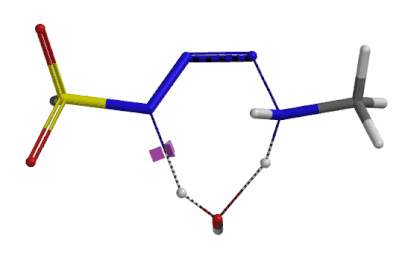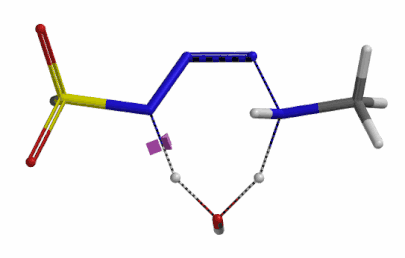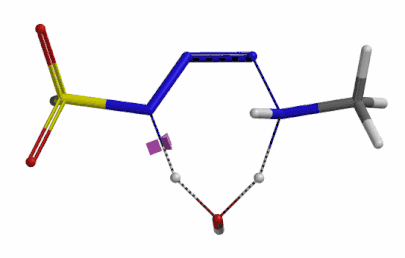
Figure 8. Transition state structures for amine addition to N-3 of FSO2N3 and hydrogen atom transfer steps.
Imaginary frequencies 136 cm-1 (left) & 1280 cm-1 (right)
Next, we calculated for the reaction energy profile of the N-1—N-2 bond cleavage of the amine-FSO2N3 addition complex and the transfer of the second hydrogen atom from amine to N-1 step, via a water molecule. Result is supportive of a concerted mechanism with an activation energy of about 20 kcal/mol (Figure 9). This step has the highest energy barrier for the whole diazo transfer reaction, indicating that this is the Rate Limiting Step of the reaction. Calculated transition state has a single ifrequency at 239 cm-1, offering further support to our water catalysis model.
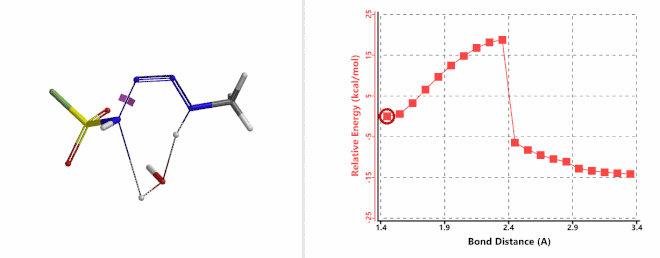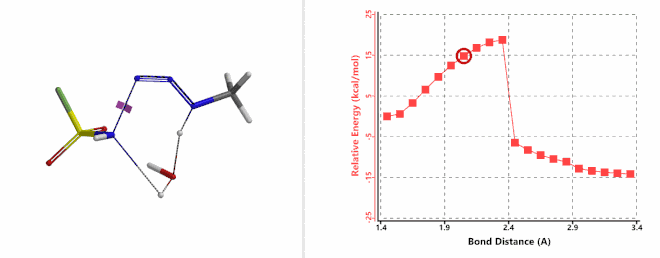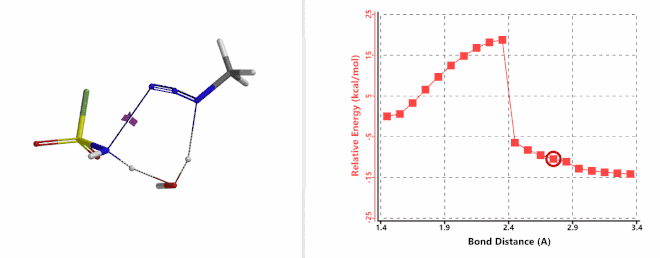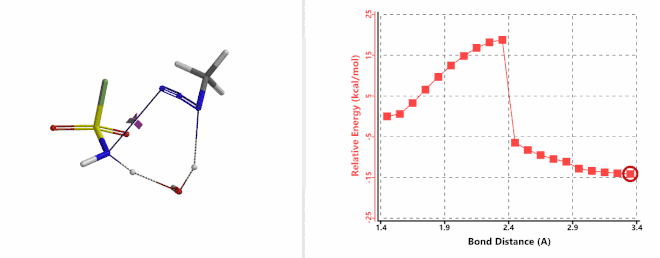
Figure 9. Reaction Energy Profile of step 2: N-1—N-2 bond of cleavage and second hydrogen transfer via water.
Shown on Figure 10 is an overall reaction energy profile of the reaction. We set the relative energy of the starting species as zero, and included the key reaction intermediates and transition states in the energy plot. The energy barrier of each step of the reaction is relatively low, consistent with room temperature reaction conditions. The reaction is exothermic, ~22 kcal/mol. Reversing the reaction from the products side will encounter an energy barrier of >30 kcal/mol, consistent with the observation that this diazotransfer reaction is not reversible at room temperature.
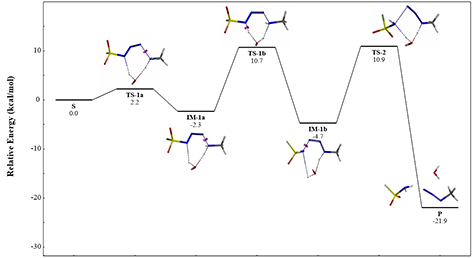
Figure 10. Overall Reaction Energy Profile of FSO2N3 diazotransfer reaction
In summary, to establish our current QM model of this FSO2N3 diazotransfer reaction, we calculated reaction energy profiles for various possible mechanisms that could account for the key experimental observations. We conducted further transition state and imaginary frequency calculations to ensure that they meet the essential criteria. A useful protocol for qualitative QM study of reaction mechanism of interest.
Reaction of carbon dioxide with water to form carbonic acid is a process of important geological and biological significances. Naturally, we are curious to find out how many water molecules are involved in the formation of a molecule of carbonic acid[6] [7] [8]. One, two, three, or four?
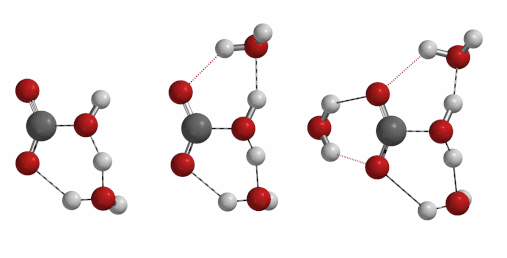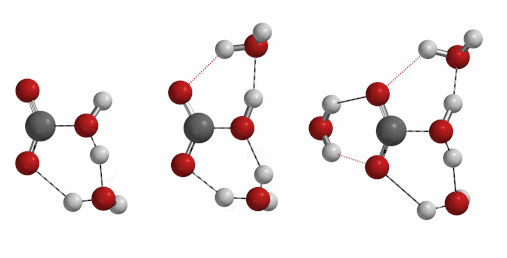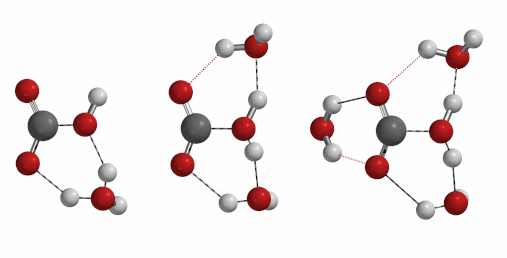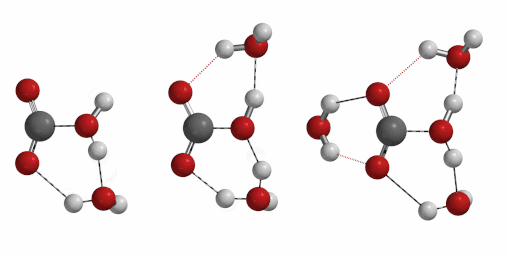
Freq 732 cm-1 iFreq 1032 cm-1 iFreq 39 cm-1 (Irrelevant vibration)
References:
[1] X.Y. Jiang, X. Hao, L.L Jing, G.C Wu, D.W. Kang, X.Y. Liu, P. Zhan, Expert Opin Drug Discov, 2019, 14, 779.
[2] G.Y. Meng, T.J. Guo, T.C. Ma, J. Zhang, Y.C. Shen, K.B. Sharpless, J.J. Dong, Nature, 2019, 574, 86.
[3] A.K. Pandiakumar, S.P. Sarma, A.G. Samuelson, Tetrahedron Lett. 2014, 55, 2917.
[4] D.C. Young, Computational Chemistry: A Practical Guide for Applying Techniques to Real-World Problems, Wiley, New York (2001).
[5] See Chapter 22, reference 5 and footnote.
[6] M.T. Nguyen, G. Raspoet, L.G. Vanquickenborne, J. Phys. Chem. A 1997, 101, 7379 (Molecular Orbital Model: water self catalysis, n = 3).
[7] R.K. Lam, A.H. England, A.T. Sheardy, O. Shih, J.W. Smith, A.M. Rizzuto, D. Prendergast, R.J. Saykally, Chem. Phys. Lett., 2014, 614, 282. (First Principles Molecular Dynamic Model and X-Ray Absorption Spectroscopy: hydration number = 3.17)
[8] S. Lindskog, Pharmacol. Ther., 1997, 74, 1. (Zn++ Catalysis, n = 1)