 选择语言
选择语言



新闻和活动

关于我们
服务和解决方案
新闻和活动
职业发展
服务平台
QM有机化学课堂
市场活动
新闻
QM有机化学课堂
在第二十五章《评估反应活性的简单方法-LUMO和HOMO的能量差中》中,我们介绍了利用LUMO/HOMO的能级能量差来快速评估相关反应活性,并根据反应位点前线轨道 (FMO) 的可用性来选择合适的轨道,计算能级差。本章则将对芳香亲核取代反应中,轨道能量的高低与反应活化能之间的相关性进行考察分析。
在《杂环化学》教材中的杂环亲核取代反应章节内,给出了一系列取代的2-氟吡啶化合物在乙醇中发生亲核芳香取代反应的反应速率常数[1]。通过将2-氟吡啶和含取代基的2-氟吡啶等当量混合,并与不足当量的乙醇钠在乙醇中反应,通过检测两个原料和对应的2位乙氧基取代的产物含量,得到各个反应底物的相对于2-氟吡啶的相对反应速率,并计算出相对活化自由能ΔΔG,具体数据见表1[2]。
由于发生的是亲核芳香取代反应,于是我们用QM计算了具有不同取代基的2-氟吡啶原料的的LUMO和LUMO+1能级,计算级别为ωB97X-D/6-31G*,其数据列于表1。我们首先使用LUMO轨道能级和实验获得的相对活化能进行作图,并对这两个变量进行线性回归分析 (linear regression analysis),其结果见图1。我们发现LUMO轨道能级和相对活化能的线性相关性并不好,其决定系数 (coefficient of determination, R2) 仅有0.5379。我们观察了2-氟吡啶原料的LUMO lobe,发现LUMO lobe主要分布在氮原子和碳碳双键上,而反应中心的2位碳原子上往往没有分布。这说明我们选取LUMO能级作为相关性分析的变量并不恰当。Galabov等人也报道了亲电芳香取代反应中存在类似的弱关联性[3]。
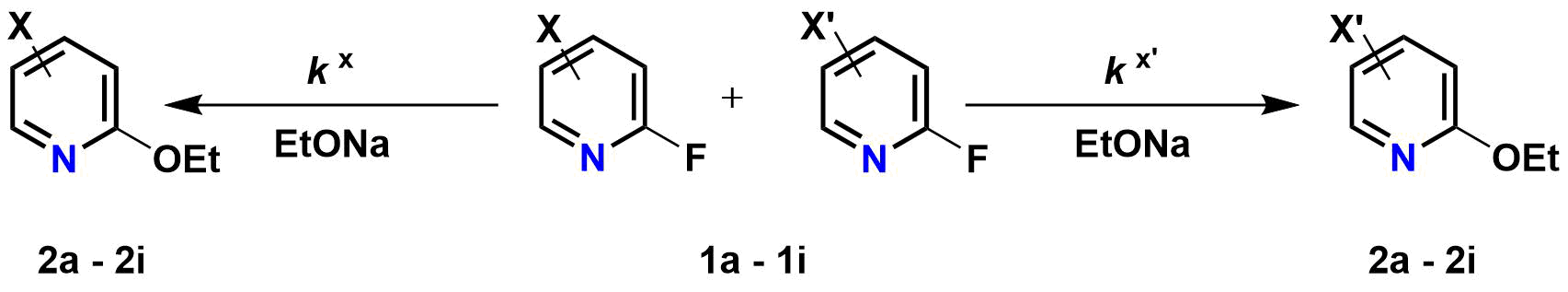
表1 不同取代的2-氟吡啶与乙氧基负离子进行亲核取代反应的反应速率,活化能及前线轨道能量(krel= 相对反应速率, kfrel= 统计校正后的相对反应速率)
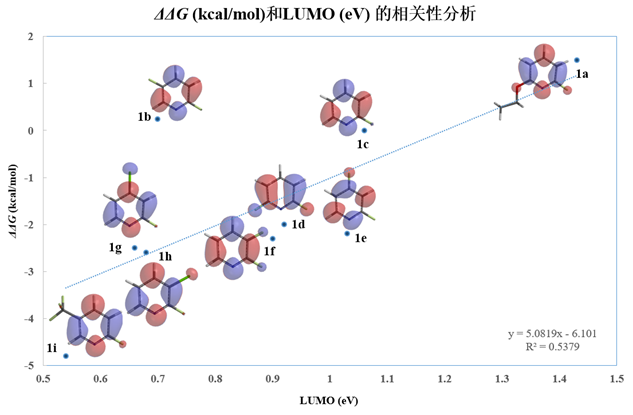
图1 不同取代的2-氟吡啶的相对活化能和LUMO能级的相关性分析
我们随后考察了取代2-氟吡啶原料的LUMO+1轨道,我们发现LUMO+1轨道和LUMO轨道分布存在显著差异,SNAr反应中心的2位碳原子上均有LUMO+1 lobe,而LUMO lobe基本不位于2位碳原子上1。
基于前线轨道理论,我们使用了LUMO+1轨道能级为变量,重新评估了它和相对活化能之间的相关性,结果见图2。我们可以看到LUMO+1和活化能具有良好的线性相关,决定系数 (R2) 显著提高到了0.9175。这说明需要选择合适的前线轨道,才能准确的根据能级大小判断反应活性的高低。
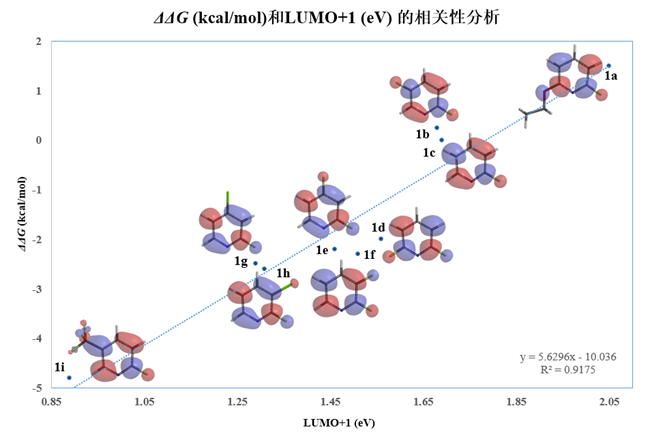
图2 不同取代的2-氟吡啶的相对活化能和LUMO+1能级的相关性分析
不同取代的2-氯吡啶的芳香亲核反应,其相对反应速率和位于反应中心位点的 LUMO/LUMO+1轨道能级值同样有着很好的线性相关,如图3所示。相关2-氯吡啶的芳香亲核取代反应数据见表2。
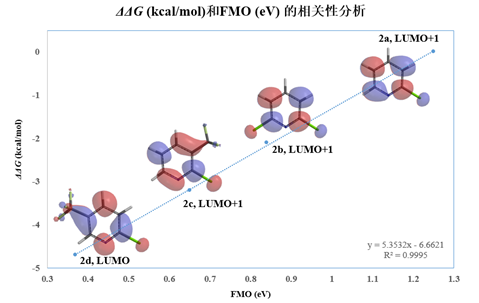
图3 不同取代的2-氯吡啶的相对活化能和FMO能级的相关性分析
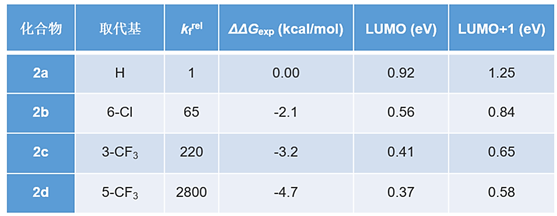
表2 不同取代的2-氯吡啶与乙氧基负离子进行亲核取代反应的反应速率,活化能及前线轨道能量
利用图3中得到的线性回归方程 (linear regression equation),我们可以利用QM计算得到的前线轨道能级,来推断对应位点的反应活性。如对于2,6-二氯-3-三氟甲基吡啶(图4),其QM计算结果表明,LUMO lobe覆盖了C-2原子,LUMO+1 lobe覆盖了C-6原子。我们将LUMO和LUMO+1的能级分别代入图3中的拟合方程,得到其相对于2-氯吡啶的相对活化能分别为-6.07 kcal/mol 和 -5.27 kcal/mol。即由LUMO主导的吡啶6位氯的反应活性要好于LUMO+1主导的吡啶2位氯,三氟甲基的位阻效应进一步抑制了吡啶2位氯的反应活性,这样的一个结果和实验测得的结果也是相一致的 (表2)。
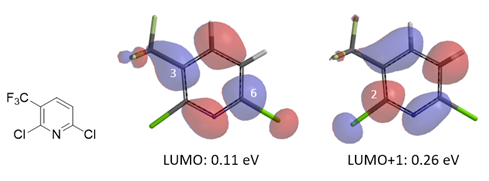
图4. 2,6-二氯-5-三氟甲基吡啶的LUMO/LUMO+1 lobe
芳香亲核取代反应受底物的LUMO能级控制,我们需要通过选择合适的前线轨道,才能够看到其能级和反应的相对活化能的良好相关性。合理选择QM计算出来的参数对于用机器学习预测反应选择性的成功与否也至关重要[4]。
在《杂环化学》教材中,作者们提到吡啶3位卤素的取代反应速率过低,没有实际用途[5]。这个说法真的能全然相信吗? 当看到下图的例子,来自用于治疗HIV-1的非核苷类逆转录酶抑制剂 (NNRTI) Doravirine的合成路线,其中酚和多取代吡啶以高产率选择性的生成了C-3位取代的产物[6]。大家都有点“尽信书不如无书”的感觉吗?

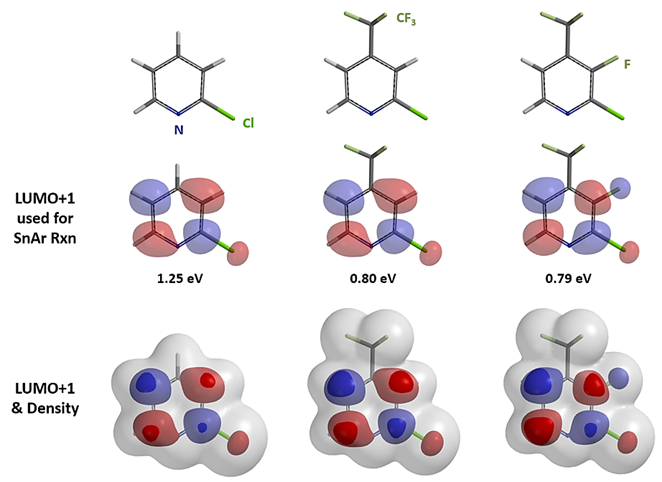
下图所示的是三个2-氯吡啶衍生物的LUMO+1及LUMO+1和电子云密度的组合图。请注意顺次引入三氟甲基和氟原子对2-氯吡啶LUMO+1能量和lobe分布的影响,以及C2和C3位点在LUMO+1和电子云密度组合图下, C3位发生亲核取代反应可行性的变化。基于这些分子轨道的信息,相信大家就能够得出上述案例中C3-F SNAr反应的选择性是完全可以预测的结论。
本文由陆颖、郑重、卫小文编撰。
参考文献:
[1] J.A. Joule & K. Mills. Heterocyclic Chemistry 5th Ed. Chichester, West Sussex, UK: Blackwell Publishing Ltd., 2010; pp 25.
[2] M. Schlosser, T. Rausis, Helv. Chim. Acta. 2005, 88, 1240-1249.
[3] G. Koleva, B. Galabov, J.I. Wu, H.F. Schaefer III, P.R. Schleyer J. Am. Chem. Soc. 2009, 131, 14722-14727.
[4] (a) T. Stuyver, C.W. Coley, J. Chem. Phys. 2022, 156, 084104. (b) Y. Guan, C.W. Coley, H. Wu, D. Ranasinghe, E. Heid, T.J. Struble, L. Pattanaik, W.H. Green, and K.F. Jensen, Chem. Sci. 2021, 12, 2198-2208.
[5] J.A. Joule & K. Mills. Heterocyclic Chemistry 5th Ed. Chichester, West Sussex, UK: Blackwell Publishing Ltd., 2010; pp 118.
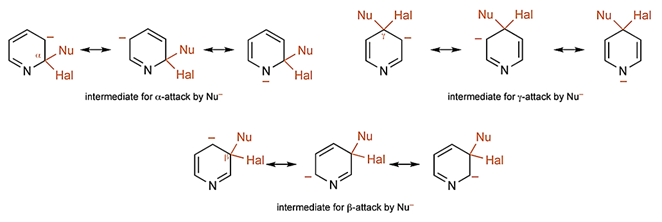
“吡啶的亲核取代反应的机理和2/4-卤代硝基苯的芳香取代反应类似,即先发生亲核试剂的加成,卤素随后离去。和苯环类底物的反应类似,加成过程受 (i) 吡啶环氮原子位和γ位的缺电子性和 (ii) 杂原子稳定加成中间体孤对电子的能力所控制。通过比较三个不同位点的加成中间体,我们可以清晰看出吡啶3位取代的中间体的负电荷无法被氮原子稳定,因此3位的取代反应速率相对大大降低——从实用角度来说可以认为它是无法发生的。”
[6] L.C. Campeau, Q.H. Chen, D. Gauvreau, M. Girardin, K. Belyk, P. Maligres, G.Y. Zhou, C.Z. Gu, W. Zhang, L.S. Tan, P.D. O’Shea, J. Org. Process Res. Dev. 2016, 20, 1476-1481.