 选择语言
选择语言



新闻和活动

关于我们
服务和解决方案
新闻和活动
职业发展
服务平台
QM有机化学课堂
市场活动
新闻
QM有机化学课堂
关于多卤素嘧啶在不同类型反应中的区域选择性,我们已经在第1章、第7章、第29章介绍过了。本章则是为大家解惑2-甲磺酰基-4-氯嘧啶在反应过程中的区域选择性问题。如图1所示,类似于2,4-二氯嘧啶的取代反应,2-甲磺酰基-4-氯嘧啶与胺的芳香亲核取代也发生在4位,但是与氧负离子的芳香亲核取代反应会选择性地发生在2位;与锡试剂做Stille偶联反应时,4位发生反应;若是将芳香胺转化为芳香甲酰胺后再做取代反应则发生在2位。
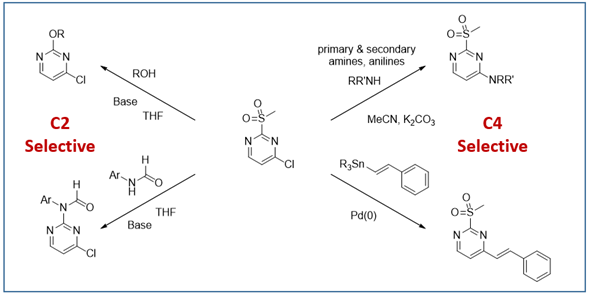
图1. 2-甲磺酸-4-氯嘧啶的SNAr和偶联反应区域选择性对比
2-甲磺酰基-4-氯嘧啶在这些反应中表现出如此大的区域选择性差异,是什么原因引起地呢?
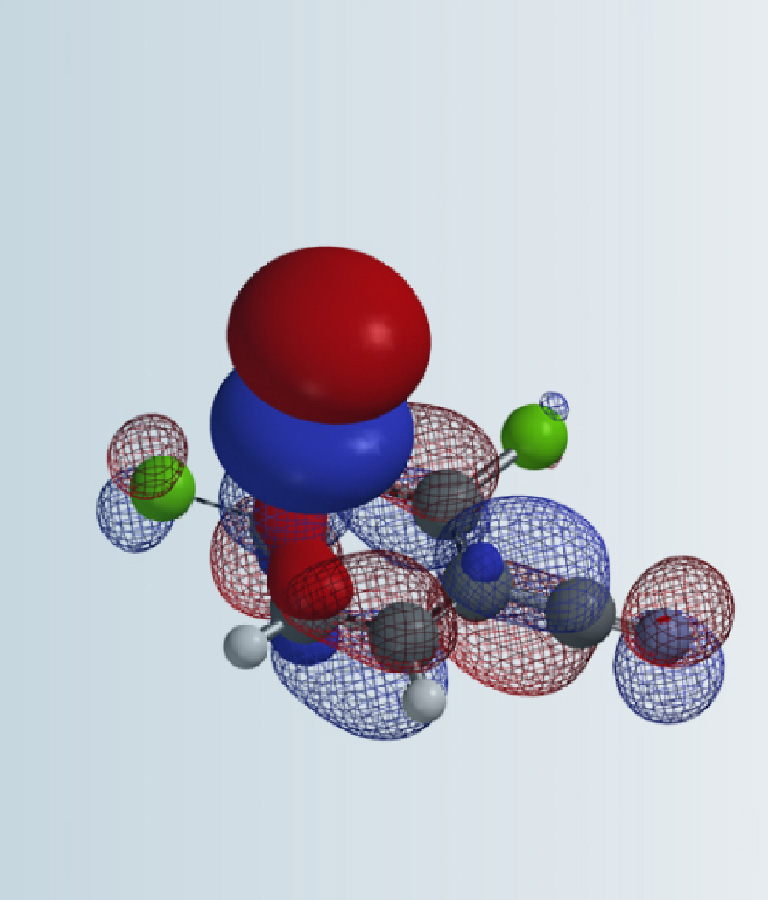
经检索发现,已有多篇文献报道了2-甲磺酰基-4-氯嘧啶与氧负离子的芳香亲核取代反应仅发生在2位,而且在-78 °C反应即可发生[1],但是从1993年第一次报道至今,我们没有检索到相关报道对这样的实验现象做出合理解释,其原因一直困扰着我们[2]。这一章将用QM推测它的反应历程,并尝试对其做出合理的解释。首先对2-甲磺酰基-4-氯嘧啶进行分析,如图2所示,C4位有LUMO分布,C2位有LUMO+1分布且能量相近,仅差0.09 eV,这表明C2和C4都可以发生取代反应。
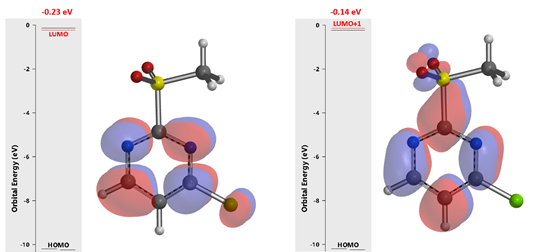
图2. 2-甲磺酰基-4-氯嘧啶的LUMO及LUMO+1示意图
我们假设当氧负离子向2-甲磺酰基-4-氯嘧啶靠近时,氧负离子与甲磺酰基的甲基氢之间有氢键作用。第一步是计算这个氢键复合物的Equilibrium Geometry。计算结果显示氧负离子与甲磺酰基的甲基氢之间是1.86 Å (图3,左),通过电子密度图(图3,中和右)可以观察到非常强的氢键作用,这可能是引导C2位发生SNAr反应的关键因素。
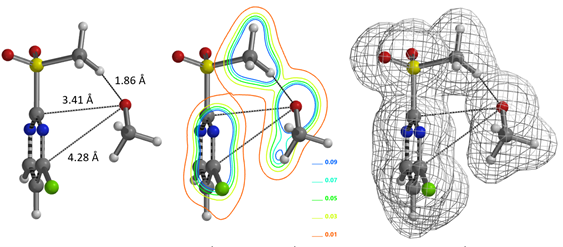
图3. 2-甲磺酰基-4-氯嘧啶与氧负离子结合的反应结构 equilibrium geometry、电子密度切片等值线图和表面示意图(计算级别:WB97X-D/6-31+G*)
先计算模拟氧负离子进攻C2原子的过程,将C2-O的距离变化设定为3.41 Å到1.51 Å (步长为0.05 Å),计算得到C2取代反应过程的能量变化曲线(见图4左侧),活化能约为0.25 kcal/mol,所以反应在-78 oC低温就很容易进行。我们可以观察到整个过程氢键是保留的,它稳定了反应过渡态并形成了一个烷氧盐加成氢键复合物。接着计算甲磺酰基在消除没有能量壁垒。然后模拟氧负离子进攻C4原子的过程,将C4-O的距离变化设定为4.28 Å到1.48 Å (步长为0.1 Å),计算得到C4取代反应过程的能量变化曲线(见图4右侧),活化能约为4.11 kcal/mol。从图4中可以看到当C4-O的距离从2.88 Å到2.78 Å,曲线有一个下降的过程,-SO2CH3的H 到 O的距离也从2.23 Å变为3.55 Å,这是氢键作用断开的过程,能量下降随后继续上升,直到C4-O成健,能量显著下降,完成C4取代反应的完整过程。C4位发生取代的过渡态能量要比C2位高3.86 kcal/mol,所以仅选择性地在C2位发生反应。此外,我们分别计算了两个过渡态的虚频,分别有且只有一个,说明这两种过渡态结构都是合理的。C2位和C4位过渡态C-O键距离分别为2.64 Å和2.38 Å。前者较长,属于早期过渡状态,而且C2的过渡态与基态相比,嘧啶的空间几何没有产生显著的变化(见图4, 5)[3]。
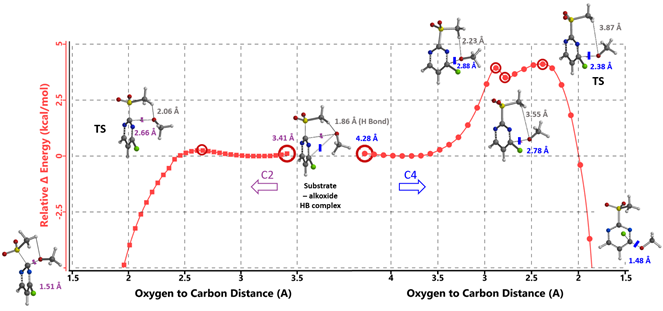
图4. C2 及 C4反应途径的能量变化曲线
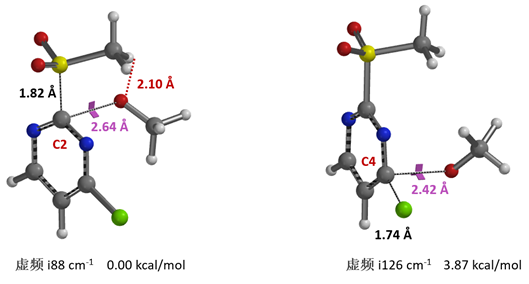
图5. 氧负离子对于在C2 及 C4位置取代下过渡态结构及虚频
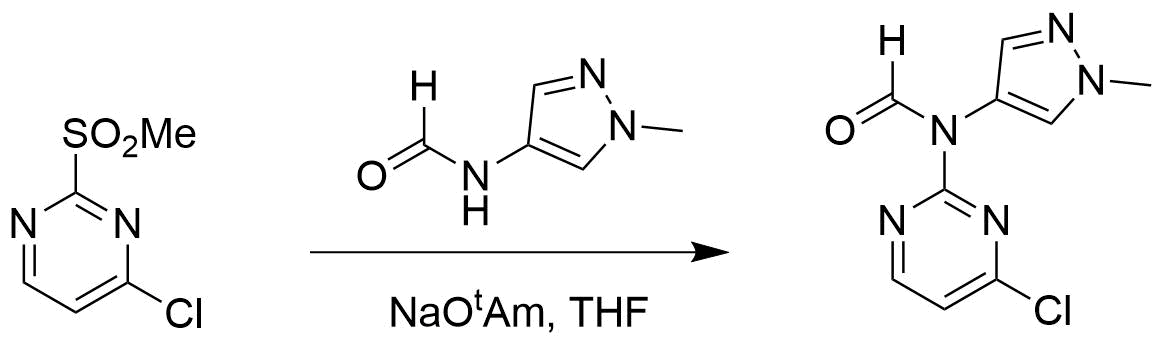
图6. 与芳香甲酰胺化合物的反应
文献报道了2-甲磺酰基-4-氯嘧啶与芳香甲酰胺的亲核取代反应也是发生在C2位[4, 5],那么它的反应机理又是怎样的呢?我们以2-甲磺酰基-4-氯嘧啶与甲基吡唑甲酰胺的反应(图6)为例,首先结合2-甲磺酰基-4-氯嘧啶与芳香甲酰胺阴离子的Equilibrium Geometry来计算它们的反应能量分布,并建立甲酰胺阴离子进攻步骤的起始模型(图7),可以看到起始的过渡态不仅在甲磺酰基的H 和甲酰胺 N有一个2.24 Å 的氢键的作用力,还存在分子间的pi-pi作用力。
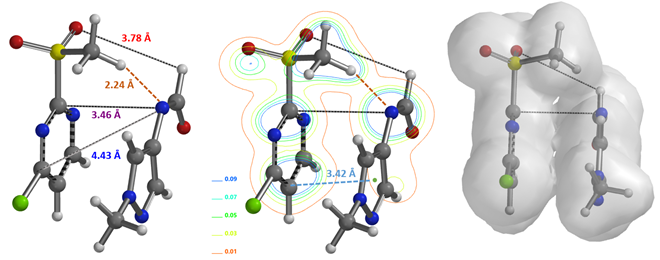
图7. 2-甲磺酰基-4-氯嘧啶与甲酰胺阴离子结合的反应结构、电子密度切片等值线图和表面示意图
模拟甲酰胺阴离子进攻C2原子的过程,计算得到C2取代反应过程的能量变化曲线(见图8左侧),活化能约为8.75 kcal/mol,反应温度需在室温到50 oC进行,整个过程氢键依然是保留的。再模拟进攻C4原子的反应过程,计算得到其能量变化曲线(见图8右侧),活化能约为14.71 kcal/mol,需要断开氢键作用力,跨越能垒相比进攻C2位要高,因此反应高选择性的发生在C2。
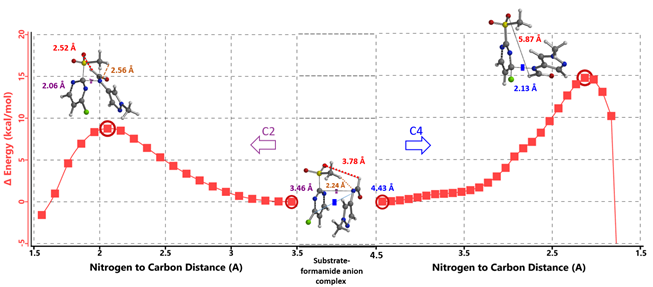
图8. C2 及 C4反应途径的能量变化曲线
我们分别计算两个过渡态的虚频以及相对能量(图9)。C4位发生取代的过渡态能量比C2位高4.12 kcal/mol,从C2位取代的过渡态结构的切片等值线图(图10)中可以看到,除了文献中提到的亲核试剂需要阴离子化来提高C2位的区域选择性的原因以外还有两个重要的原因:1)-SO2CH3 H 对甲酰胺 N 的氢键(2.58 Å)诱导甲酰胺离子进攻C2原子。2)氢键会在甲酰胺的氢和MeSO2-氧之间形成 (2.53 Å),进一步稳定了过渡态,降低了活化能,这就解释了为什么只有甲酰胺可以提高分离收率和选择性。甲酰基在这个高选择性的反应中起着关键作用。
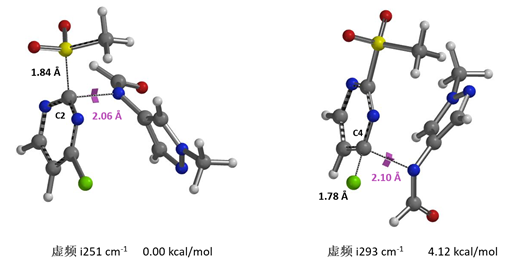
图9. 甲酰胺阴离子对于在C2 及 C4位置取代下过渡态结构的虚频及相对能量
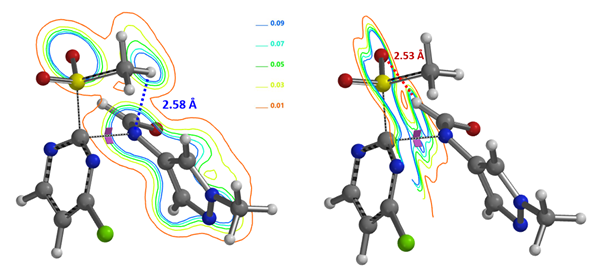
图10. 甲酰胺阴离子对C2位取代下过渡态结构的电子密度切片等值线图
通过上面的两个QM的例子,我们可以给出以下结论:
在C2位的取代反应的原因有:1. 阴离子化的亲核试剂与甲磺酰基的质子存在氢键作用;2. 氢键可以稳定反应过渡态;3. 甲磺酰基会引导阴离子化的亲核试剂去进攻C2位原子从而实现了C2位的芳香取代反应。
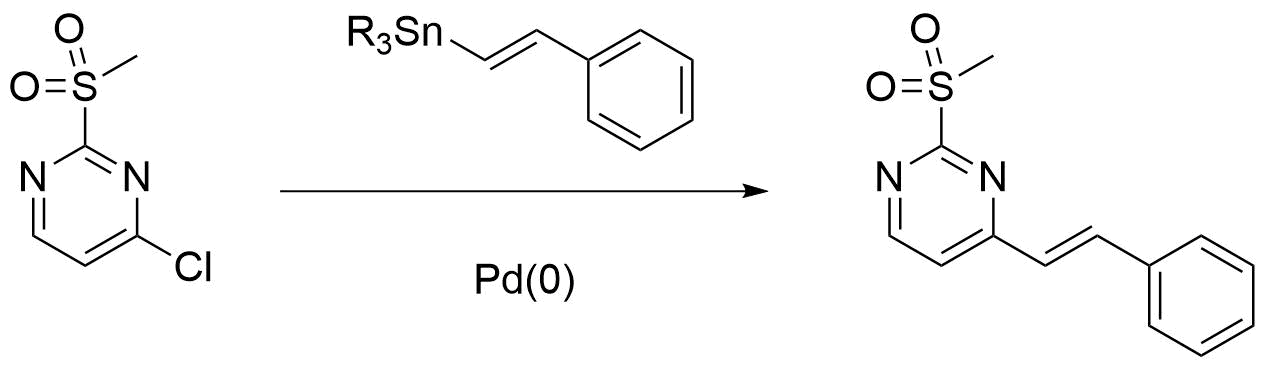
图11.在Stille偶联反应中的区域选择性
那么为什么氧化加成反应会发生在C4位呢?Recently, Chatelain et al [6] reported苯基砜芳基砜在Pd(0)催化下偶联Suzuki反应中表现出介于芳基氯化物和硝基芳烃之间的反应活性(图11),以及三氟甲基苯砜反应收率最好。此时,我们可以给出结论:在氧化加成反应过程中,芳基上Cl的活性远大于甲磺酰基,Pd(0)更容易与C-Cl键发生氧化加成,所以偶联反应发生在C4位。
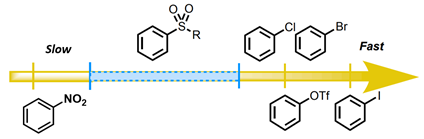
图12. 芳香亲电试剂在Suzuki反应中的相对反应活性差异图
2-甲磺酰基-4-氯嘧啶在进行芳香亲核取代反应时甲磺酰基具有诱导效应,与阴离子化的亲核试剂产生的氢键相互作用,可以稳定其过渡态并可能阻断潜在的反应途径。当反应涉及氢键、非共价相互作用(Non-Covalent Interaction, NCI)[8],空间相互作用和电子相互作用等时(见第 9, 23, 26, 33, 42章),它们的反应结果可能会变得很复杂,我们往往无法直观的预测。QM是一个学习有机化学的强大工具,一个客观分析方法。基于实验的条件和现象来学习建立及不断改进计算反应模型,我们就可以加深对反应的理解。如果你们遇到了其他有趣的化学,请告知我们,大家一起解析学习。
2-甲磺酰基-4-氯嘧啶与仲胺反应仅发生4位取代,这又要如何分析呢?。
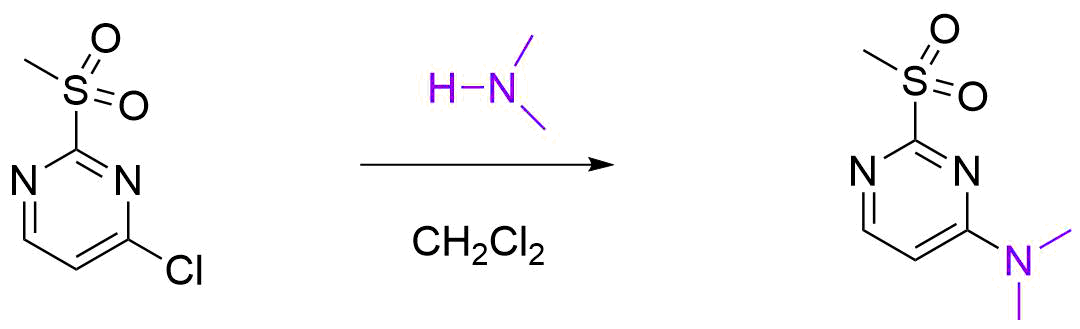
图13. 2-甲磺酰基-4-氯嘧啶与胺反应
二级胺,其N原子上的孤对电子和氢原子,有多种可能的方式与MeSO2基团和嘧啶的sp2 N相互作用。计算表明它们形成一个复合物,ΔE为-7.61 kcal/mol(结构如图14所示)。电子密度等值线图揭示:
1. 二级胺氮原子(孤对电子)与亲电性嘧啶C-2碳之间有强非共价作用(2.98 Å)。
2. 胺氮和MeSO2基团氢之间存在一个氢键(2.55 Å)。所展示的复合物结构比评估的其他胺类H与MeSO2氧氢键键合的结构能量更低(> 1千卡/摩尔)。
3. 二级胺甲基基团上的氢原子与嘧啶N-1和N-3之间存在两个电荷-电荷相互作用(自然电荷)。
您会如何进一步分析这个反应?[9]
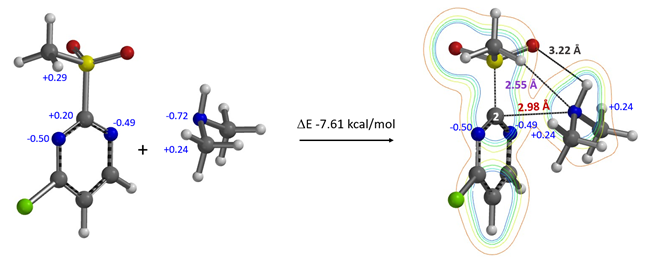
图14. 2-甲磺酰基-4-氯嘧啶与仲胺形成复合物的形成; 电子密度切片等值线图及自然电荷(蓝色标记)
本文由陆颖、刘文锋、王秋月、卫小文编撰。
参考文献:
[1] M. L. Falck-Pedersen, B. Tore, U. Kjell, Acta Chem. Scand. 1993, 47, 72.
[2] C. M. Serrano, R. E. Looper, Org. Lett. 2011, 13, 5000; A. S. Carlson, H. R. Cui, A. Divakaran, J. A. Johnson, R. M. Brunner, W. C. K. Pomerantz, J. J. Topczewski, ACS Med. Chem. Lett. 2019, 10, 1296.
[3] F. M. Bickelhaupt, K. N. Houk, Angew. Chem. Int. Ed. 2017, 56, 10070. Small HOMO and LUMO energy gap and little structure distortion from reactant to transition state leads to low activation energy.
[4] C. M. Li, F. Haeffner, S. J. Wang, C. C. Yuan, D. Shang, X. L. Shi, B. Ma, B. T. Hopkins, E. M. O’Brien, Org. Process Res. Dev. 2022, 26, 137.
[5] B. Barlaam, R. Ducray, C. Lambert-van der Brempt, P. Plé, C. Bardelle, N. Brooks, T. Coleman, D. Cross, J. G. Kettle, J. Read, Bioorg. Med. Chem. Lett. 2011, 21, 2207.
[6] P. Chatelain, A. Sau, C. N. Rowley, J. Moran, Angew. Chem. Int. Ed. 2019, 58, 14959.
[7] B. L. Mylari, P. J. Oates, W. J. Zembrowski, D. A. Beebe, E. L. Conn, J. B. Coutcher, M. T. O'Gorman, M. C. Linhares, G. J. Withbroe, J. Med. Chem. 2002, 45, 4398.
[8] QM Chapter 42
[9] To continue with the analysis, we’ll use the (global minimum energy) structure obtained for 2-MeSO2-4-Cl-pyrimidine-secondary amine complex (shown in figure 14) to set up C-2 and C-4 SnAr REP calculations. Then it will become obvious that the energy barrier of the C-4 substitution is lower than that for C-2. To understand why, transition state calculation, followed by activation strain (distortion) analyses[3] will be very informative. They reveal that the C2- transition state is associated with a larger strain energy, the key reason why SnAr with secondary amine is C-4 selective.