 选择语言
选择语言



新闻和活动

关于我们
服务和解决方案
新闻和活动
职业发展
服务平台
QM有机化学课堂
市场活动
新闻
QM有机化学课堂
我们在本课中将讨论3-乙酰基-5-羟基吲哚1亲电反应的区域选择性问题。如图1所示,在对吲哚底物1进行溴化时只单一得到6位溴化的产物2,并没有观测到4位溴化产物2A的生成。但其参与的Mannich反应却具有高度C4选择性[1]。如何解释这种选择性差异?
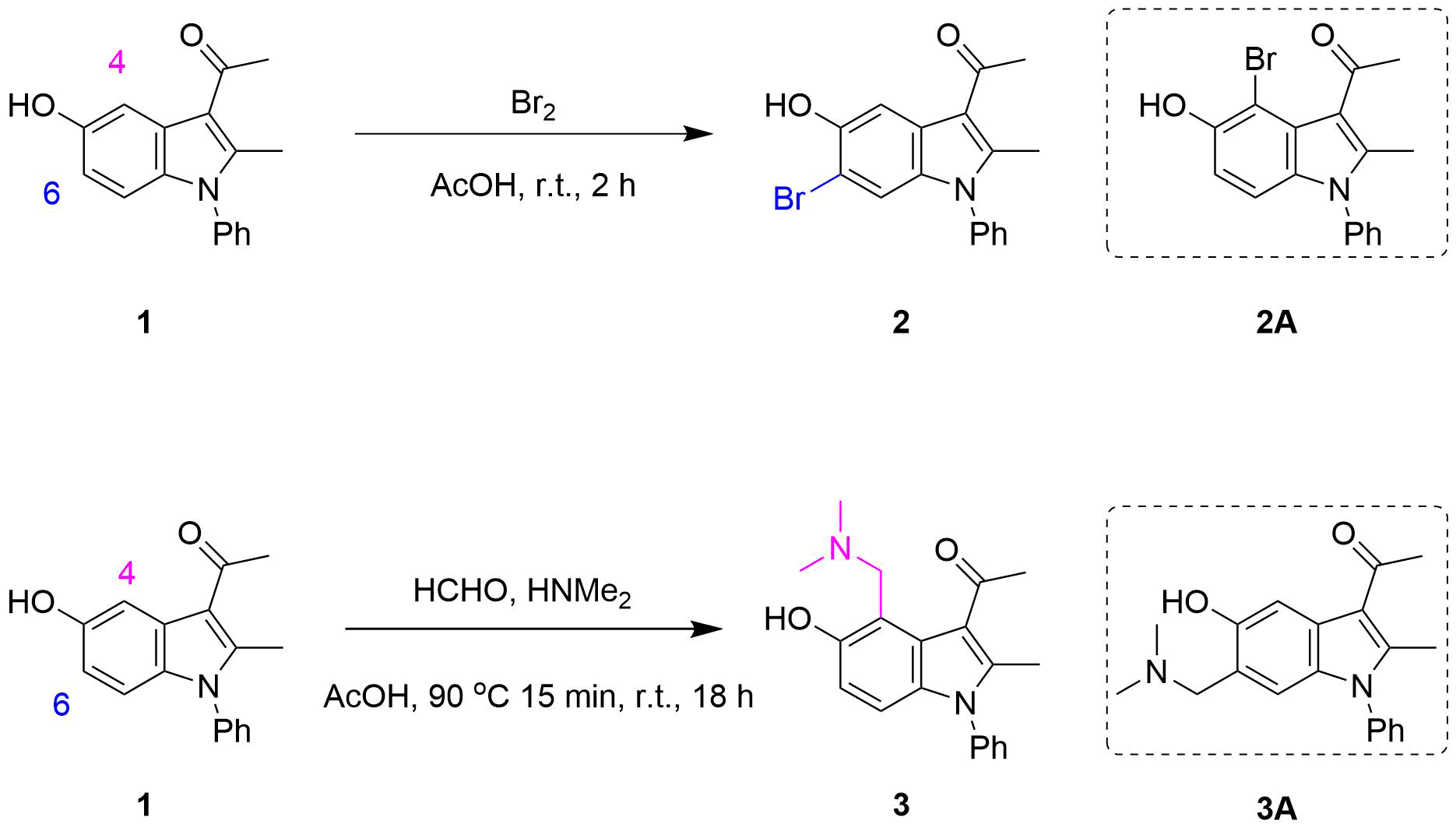 图1. 3-乙酰基-5-羟基吲哚1的溴代与Mannich反应
图1. 3-乙酰基-5-羟基吲哚1的溴代与Mannich反应
首先,我们先考察化合物1的溴化反应。在第二章《亲电反应中HOMO计算的运用》中,我们可以通过计算底物可能反应位点的HOMO lobe大小和13C NMR来预测反应的选择性。如图2所示,计算结果表明HOMO (-7.46 eV) 及HOMO-1 (-7.57 eV) 能量相近,C4的HOMO lobe和C6的HOMO-1 lobe相差不大,选择性不会很好,同时C4的13C化学位移数值更低,C4位取代应该比较多。显然计算结果和实验数据不相符。
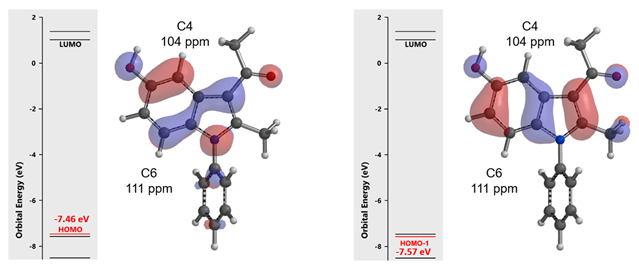
图2. 吲哚化合物1的HOMO, HOMO-1及计算 13C NMR 示意图示意图
我们在第十七章《亲电卤代反应的区域选择性预测方法》中介绍过通过计算相关的卤代正离子平衡构象以及相对能量来预测优势反应位点。我们用ωB97X-D/6-31G*计算级别对可能的溴离子平衡构象进行计算(C4考察两种构象)。计算结果显示,C4位和C6位溴代正离子的相对能量均较低,理论上应该得到以C4位为主的两个异构体,显然和实验结果也不符合。
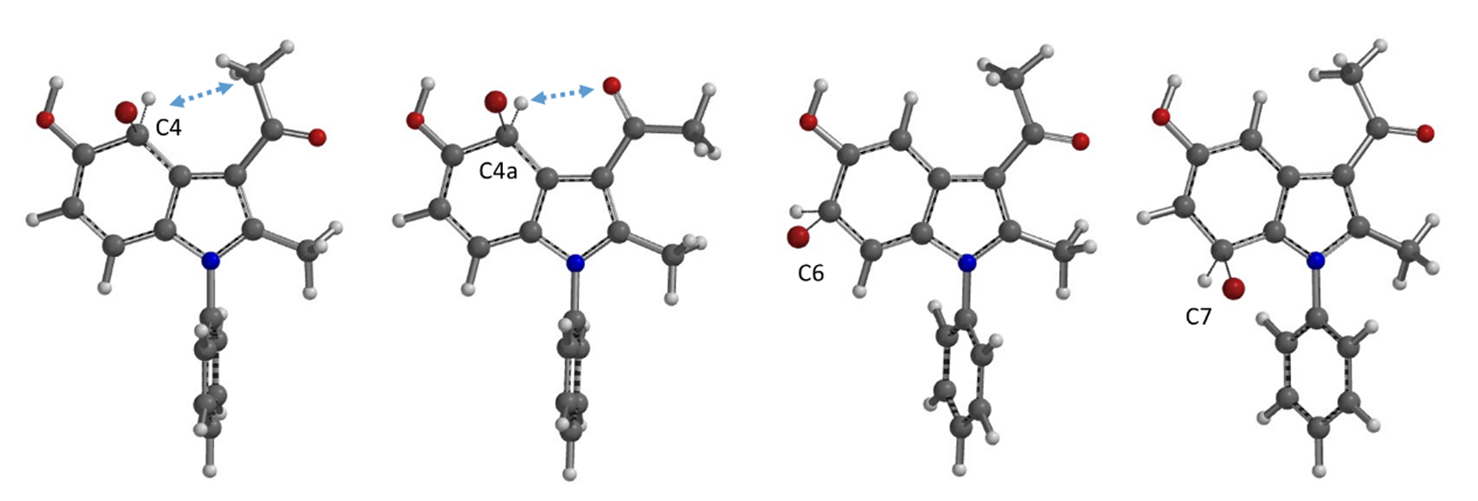
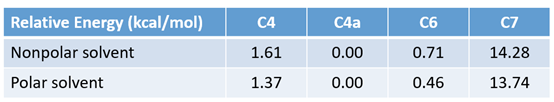
图3. C4、C4a、C6、C7位溴代正离子的平衡构象以及相对能量
那么为什么反应位点的HOMO lobe大小,13C NMR位移以及正离子平衡构象以及相对能量来预测Br2 溴化反应位点等方法在这个底物上不适用呢?我们看一下芳香化合物的亲电取代机理(图4),首先芳香环攻击亲电试剂E+,生成一个带正电荷的离域环己二烯基阳离子,也被称为Wheland中间体或芳烃σ-络合物[2],随后失去一个质子得到芳香性的产物。通常形成σ-络合物是一个决速步,随后即可快速生成产物。但是当受到某些因素如位阻影响时,会导致最后去质子化过渡态能量差成为主要决定因素。用我们的化学直觉来判断上述预测方法可能不适用,我们需要进一步计算溴化过程中每个步骤的详细情况。
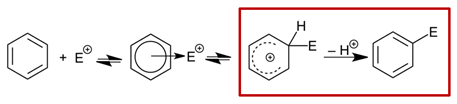
图4. 芳香化合物的亲电取代机理[2]
我们改用Br2来设置吲哚化合物1的溴化计算模型,同时计算了形成Wheland中间体后经过Br- 辅助质子离去过程的能量变化。如图5所示,在C4位取代时,Wheland中间体形成后,Br- 稳定了该中间体,这时整个体系能量最低,下一步Br- 辅助质子离去时[3],由于3-乙酰基带来的空间位阻影响,需要跨越一个11.59 kcal/mol能垒的TS2,该能垒甚至比回到原料还要高。
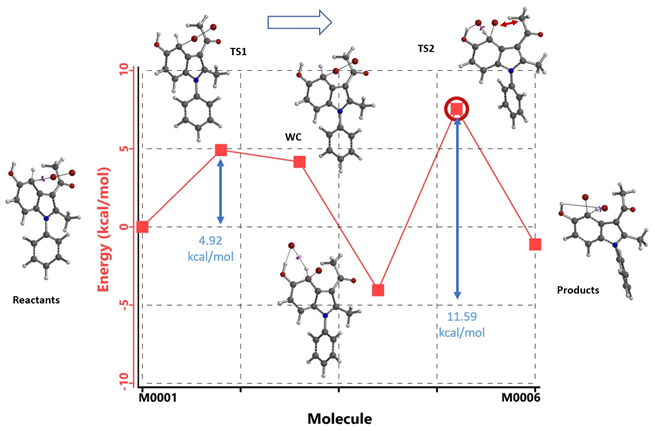
图5. 吲哚化合物1 C4溴代能量变化
在C6位取代计算模型中(图6),Br- 辅助质子离去时,没有显著的空间上的相互作用,仅需跨越一个6.71 kcal/mol能垒的TS2。因此比C4 TS2 能垒11.59 kcal/mol低4.88 kcal/mol,足以解释实验观察到的C6位溴化反应的高度选择性情况
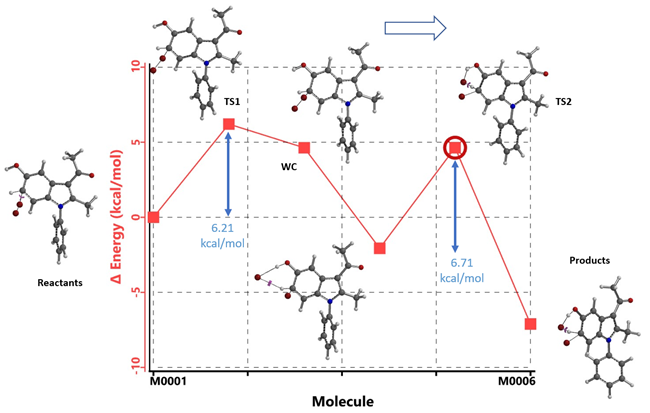
图6. 吲哚化合物1 C6溴代能量变化
综合起来,一个更贴切的计算模型 (用Br2替代Br+,同时考察去质子化/重芳构化的过程) 揭示了吲哚化合物1尽管C4位溴化形成Wheland中间体的能量比C6位略低,但是随后到终产物的过程中需要翻过一个更高的能垒。因此,此路径对C4位溴代不利,最终只得到单一C6位的溴代产物。
对于3-乙酰基-5-羟基吲哚1,Mannich 反应基本只得到C4反应产物,与溴化反应在C6位的高度位置选择性完全相反[1]。
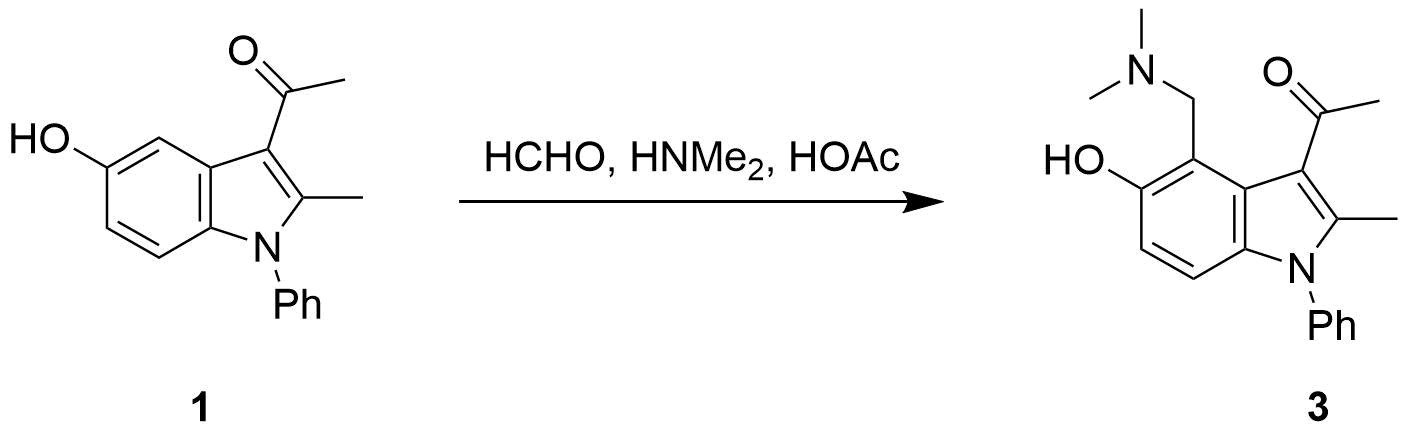
图7. 3-乙酰基-5-羟基吲哚1的Mannich反应
在Mannich反应中,甲醛和二甲基胺反应生成高活性的Mannich试剂(LUMO:-5.24 eV),从图8可知亚胺碳上的LUMO lobe明显大于N原子的LUMO lobe,这解释了化合物1对亚胺碳的选择性亲核进攻 (图8 左)。同时我们也可以计算得到Mannich试剂的自然电荷分布情况(图8 中) N原子为负电荷,甲基H为正电荷(教科书通常错误的将N标记为带正电荷[4])。与二甲胺中N原子电荷-0.698相比,Mannich试剂的N负电荷减少到-0.275,并不是带正电荷。这与Chapter 23章中提到的四甲基铵情况相似。(图8 右)[5]。
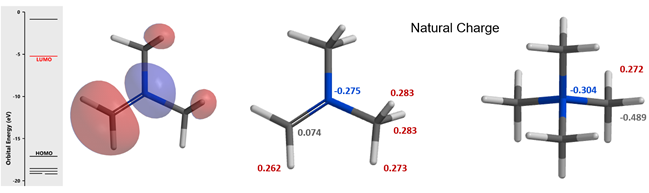
图 8. 曼尼希试剂的LUMO示意图(左)、自然电荷分布示意图(中)、四甲基铵的自然电荷分布示意图(右)
从Mannich反应发生在C4及C6位进行计算可知,当C4发生反应时,二甲胺的甲基正电荷H与底物乙酰基的C=O负电荷O存在非共价相互作用(图8 左),从而稳定过渡态, 相反在C6位反应则不存在氢键稳定作用(图8 右)。这导致两个过渡态结构能量差达到1.74 kcal/mol,Mannich反应优先得到C4反应的单一产物。
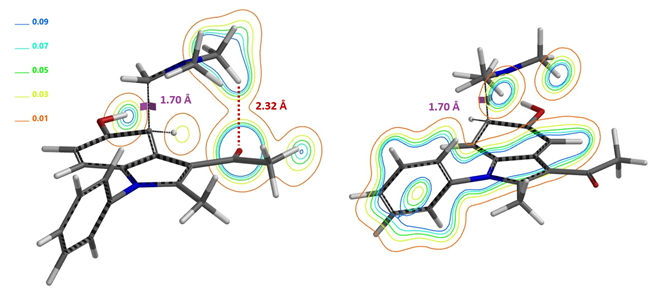
图9. C4 (左)与C6 (右) 位Mannich的反应结构及电子密度等高线示意图
同一底物发生不同类型的亲电反应,位置选择性可能不同。对于本文的吲哚底物,C4位溴化反应存在3-乙酰基带来的位阻效应,然而在Mannich反应中3-乙酰基与亚胺离子的电子效应有助于反应发生,导致不同的位置选择性。我们需要根据反应机理、潜在的邻基参与效应以及具体反应条件,设置对应的计算模型,考察合适的参数来分析理解实验现象、预测反应结果。
如图10所示,8-甲氧基喹啉(4)在NBS/CHCl3的条件下只生成5-溴化产物5,这与HOMO,13C-NMR,C5/C7位溴化正离子相对能量计算结果匹配。然而在同样条件下其类似物8-羟基喹啉(6)则生成C-5及C-7位反应的混合产物,且以C-7位溴化产物7为主,这与我们按常用参数设置计算的结果不符。
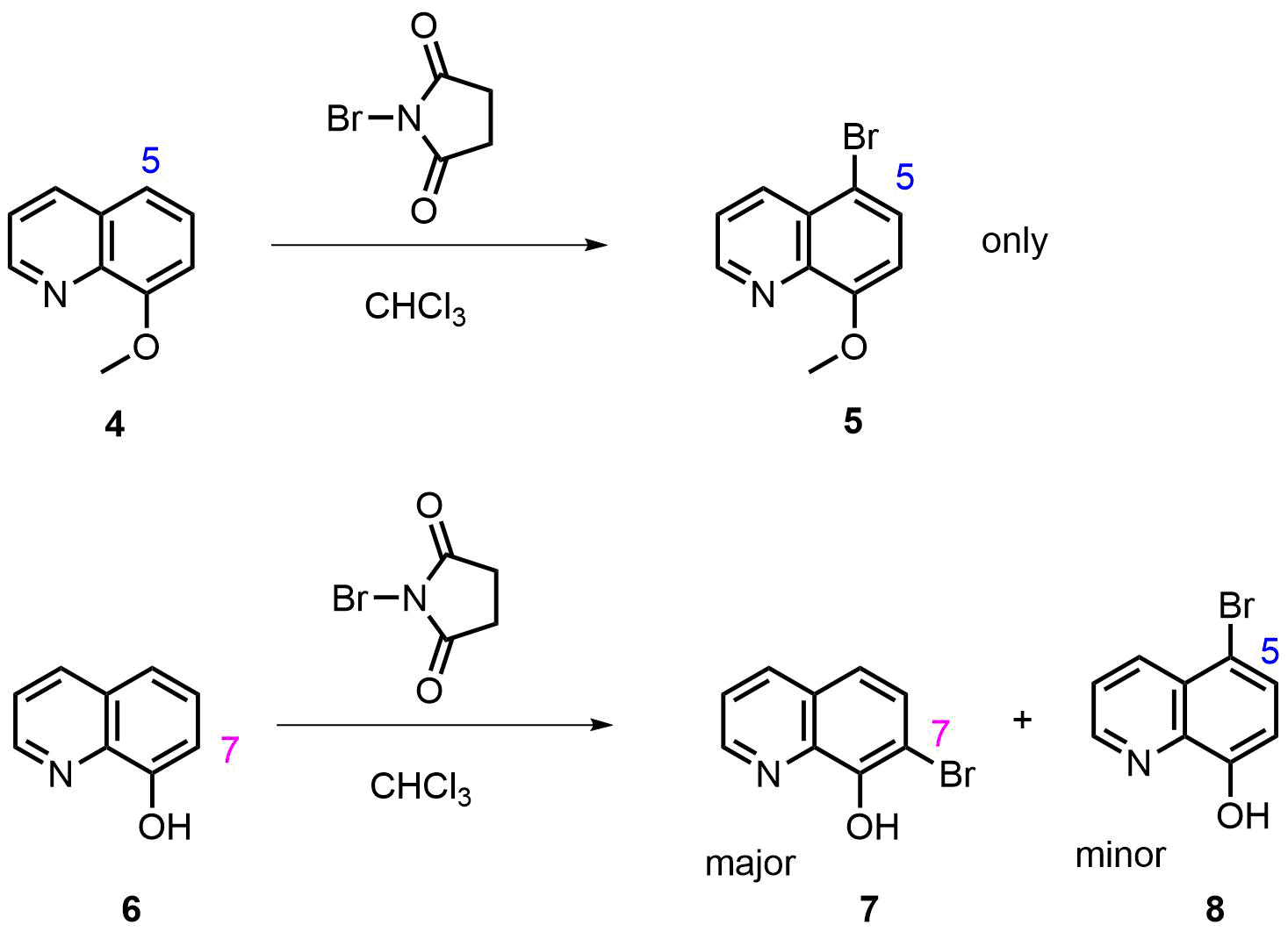
图10. 8-甲氧基喹啉与8-羟基喹啉的溴化反应(NBS为溴化试剂)
随后我们考察OH与溴化试剂间存在的潜在氢键作用来理解实验结果。进一步的QM分析也支持我们提出以下的假设:C-5 与 C-7 发生溴化反应的相对总速率由 C-5 和 C-7 位Wheland 中间体去质子化速率控制。琥珀酰亚胺与C-7位Wheland中间体的氢键作用(图11), 增加了琥珀酰亚胺去质子化步骤的有效浓度,使得去质子化更容易进行得到芳构化产物。

图11. 8-羟基喹啉-NBS复合物在HO=CN共平面时的电子密度等高线示意图
那么问题来了,如果以上论断都是正确的,用NBS作为溴代试剂与8-羟基喹啉在不同浓度下进行反应,结果如何?如果想得到C-5高度选择性的产物要如何实现呢[5]?
本文由刘俊、乔建珲、王秋月、董立亭、卫小文编撰。
参考文献:
[1] G.S. Gadaginamath, A. G. Kamat, B. G. Pujar, Revue Roumaine de Chimie, 1995, 40, 265.
[2] https://en.wikipedia.org/wiki/Electrophilic_aromatic_substitution.
[3] W.J. Hehre, A.J. Shusterman, J.E. Nelson (1998) The Molecular Modeling Workbook for Organic Chemistry. Irvine, CA, USA: Wavefunction, Inc.; pp 86-7. Sn2反应与去质子化之间的相似性。

图12. Br-从Cl-反向进攻HCl 氢及CH3Cl 碳,置换出Cl- 过程示意图
[4] E.V. Anslyn, D.A. Dougherty (2006) Modern Physical Organic Chemistry. New York, NY, USA: University Science Books; page 7.
[5] QM Chapter 23 “A QM Study of the para Regioselectivity of TBABr3 Bromination”

图13. 反应结合物C-Br距离2.6 Å中时的电子密度(Isovalue 0.00744 e/au3, 98.0%)示意图