 选择语言
选择语言



新闻和活动

关于我们
服务和解决方案
新闻和活动
职业发展
服务平台
QM有机化学课堂
市场活动
新闻
QM有机化学课堂
含氟有机化合物对药物开发具有很高的价值[1],脱氧氟化反应是将醇转化为脂肪族氟化物的重要方法。目前已经发展了多种脱氧氟化试剂,其中最常用的是DAST,但DAST热稳定性差且易爆炸。近年来发展了一系列新型的脱氧氟化试剂,如: PyFluor [2]和AlkylFluor[3]。同时碱的选择在该反应中也非常重要,如PyFluor和DBU是链状仲醇脱氧氟化反应的最佳组合,能最大程度减少消除副产物的生成[2]。
如何在众多脱氧氟化条件中,快速的寻找到最优反应条件呢?2018年,普林斯顿大学的Abigail G. Doyle教授报道,将Quantum Mechanics(QM)和机器学习(Machine Learning:ML)相结合,构建一个可靠的QM-ML模型,可以快速预测脱氧氟化反应的最优条件[4]。
经过之前多篇QM微信文章的学习,大家已经相当熟悉QM了。那什么是机器学习(ML)呢?
ML是计算机科学、数学和统计学交叉领域的子领域,可以从数据中自动分析获得规律,并利用规律对未知数据进行预测。图1显示了建立这个QM-ML模型用于预测的流程[5]。首先,收集640个筛选反应的相关数据,包括实验收率以及反应中使用的醇、磺酰氟和碱的特征数据。这些特征数据包括QM计算的原子和分子特性(即静电荷、LUMO和 HOMO 能量、碳谱数据、偶极矩等)以及反应物醇的分类信息(即一级、二级、三级、环状、苄基、烯丙基、高苄基、高烯丙基等)。然后把70%的数据作为训练集应用到随机森林算法中,得到ML模型。再使用剩余30%的数据作为测试集评估模型。验证通过的QM-ML模型可以准确预测剩余部分底物的脱氧氟化反应最优条件[4a]。
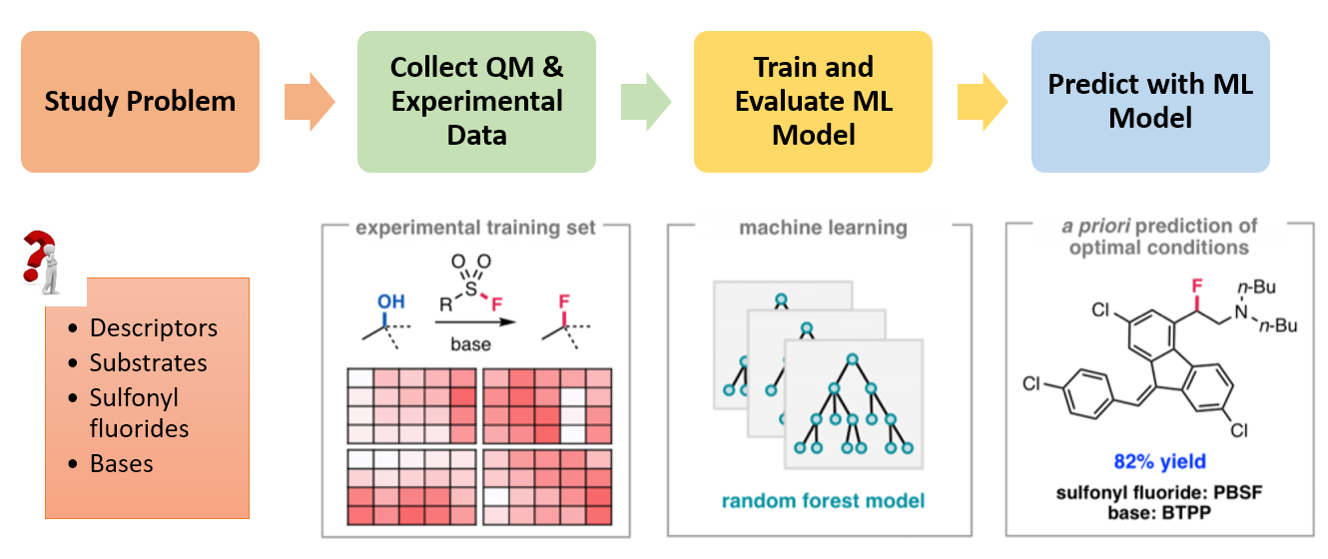
图1. 建立QM-ML模型的流程[4, 5]
图2是应用QM-ML脱氧氟化反应模型进行预测的流程。首先通过QM计算出醇类反应底物的HOMO和LUMO能量,以及与羟基相连碳的静电电荷和暴露面积。将以上4个数据以及醇的分类信息输入到QM-ML模型中,仅需要几秒钟,就可以预测底物在20个不同反应条件下的收率。一个相对简单的预测流程!
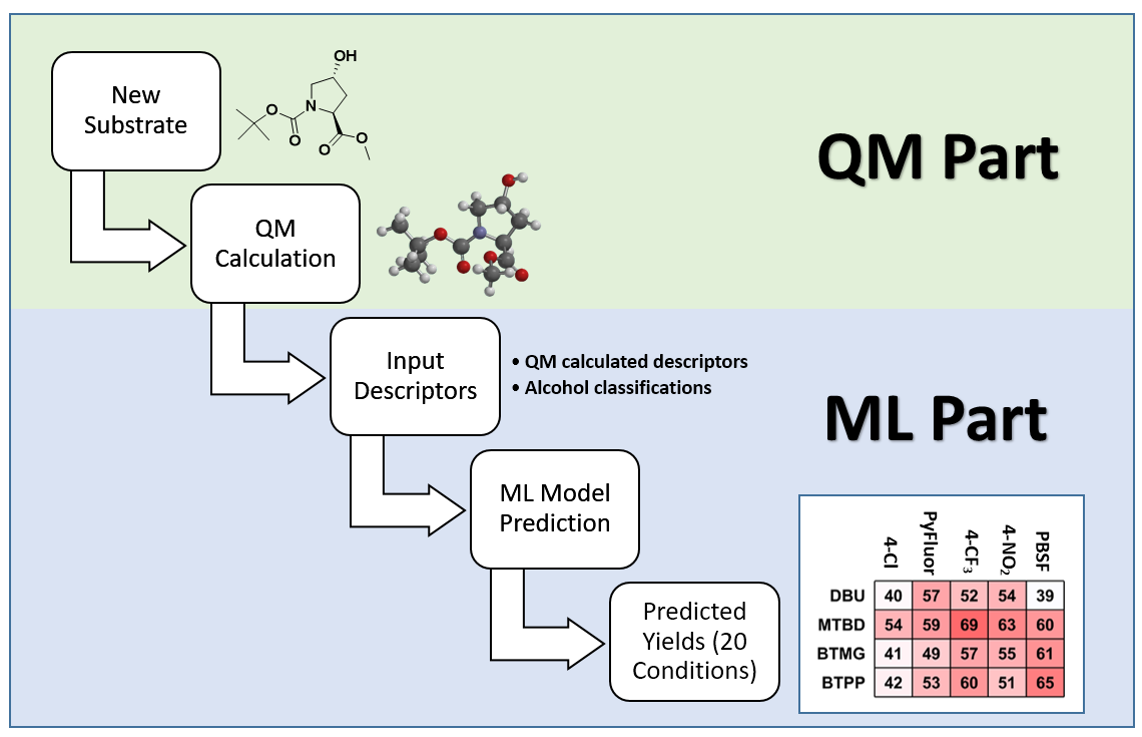
图2. 使用 QM-ML 模型进行脱氧氟化反应预测[4b]
Doyle课题组的论文发表后,因为我们已经拥有了QM计算能力,所以实验室能立即使用这个QM-ML脱氧氟化模型来寻找更好反应条件。如图3所示,化合物1中的醇羟基使用甲磺酸酯活化后,TBAF取代得到氟化产物2,反应收率为43%, 该反应损失了超过一半的珍贵中间体1。

图3. 氟化反应案例
通过QM-ML脱氧氟化模型计算,给出了如下20种条件的预测结果。最优条件为PBSF和BTPP的组合,预测收率为42.3%。
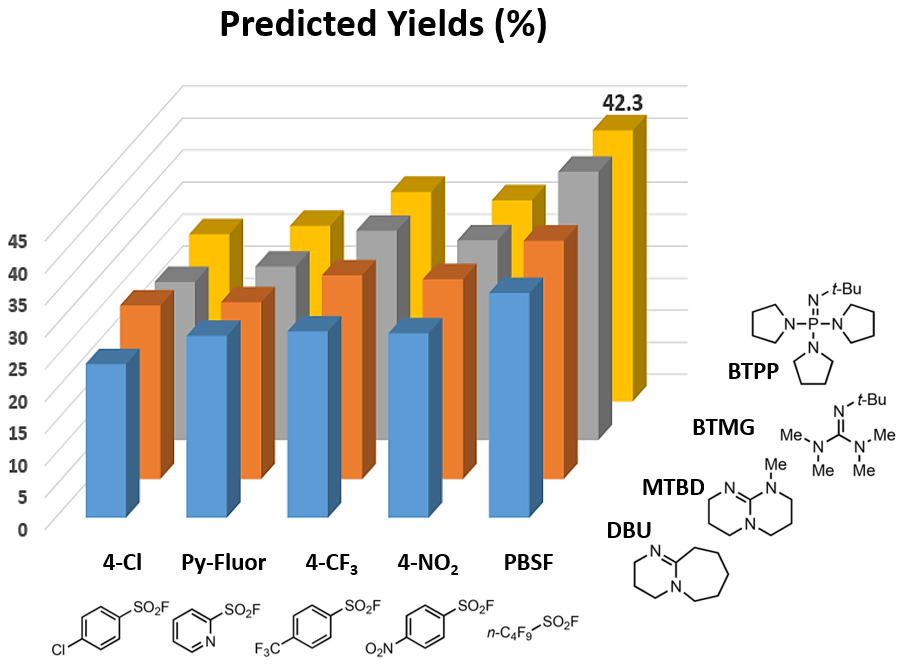
图4. QM-ML脱氧氟化反应模型预测最优反应条件[4b]
利用这个推荐的PBSF和BTPP条件,实验得到分离收率为83%,与之前的条件相比较收率提高近一倍。这是我们实验室应用QM-ML脱氧氟化反应指南的第一个案例。

图5. 最优反应条件实验结果
Doyle课题组通过QM-ML脱氧氟化模型为30多种醇找到了最优反应条件,包括活性醇(苄醇,烯丙醇),非活性的醇,链状醇,环状醇等。对于非环状的二级醇,如化合物3,ML预测的最优条件是4-CF3PhSO2F和MTBD的组合,收率为69%。但实验的最优条件是PyFluor和DBU的组合[2, 4]。所以ML预测的结果和实验情况有时会存在误差。
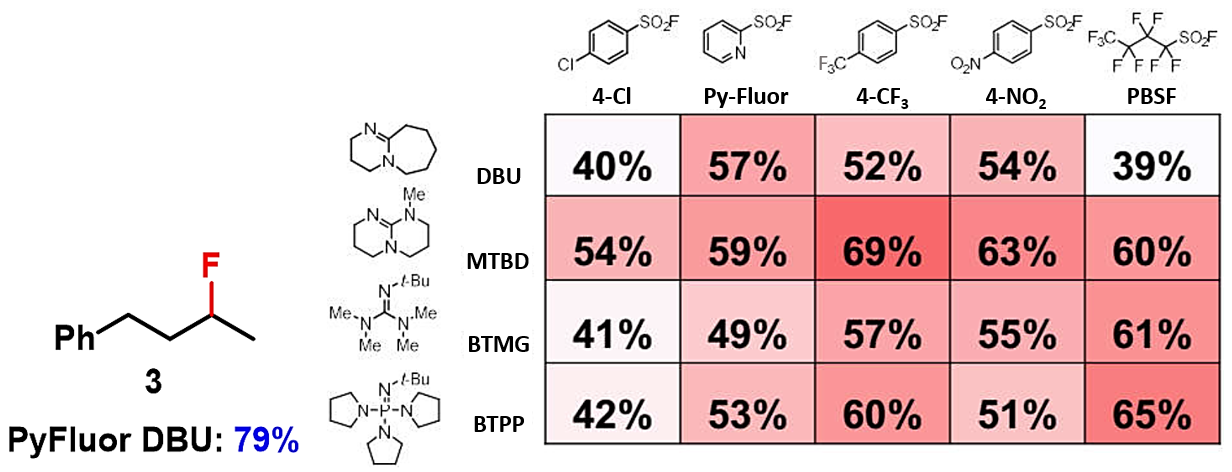
图6. 脱氧氟化反应案例
总结一下,QM和ML强强联合成功构建了准确预测脱氧氟化反应收率的模型,可以帮助我们快速找到最优反应条件。希望我们这种成功的经验也能应用到其它类型的反应中去。
我们同样提供脱氧氟化反应预测服务(图7),如果您有QM-ML脱氧氟化的计算需求,请联系我们:IDSU_Operation@wuxiapptec.com。只需提供底物结构,我们就能帮助您预测最优条件。在应用此条件后,也请把反应收率反馈给我们,这将对继续优化ML模型有很大帮助。
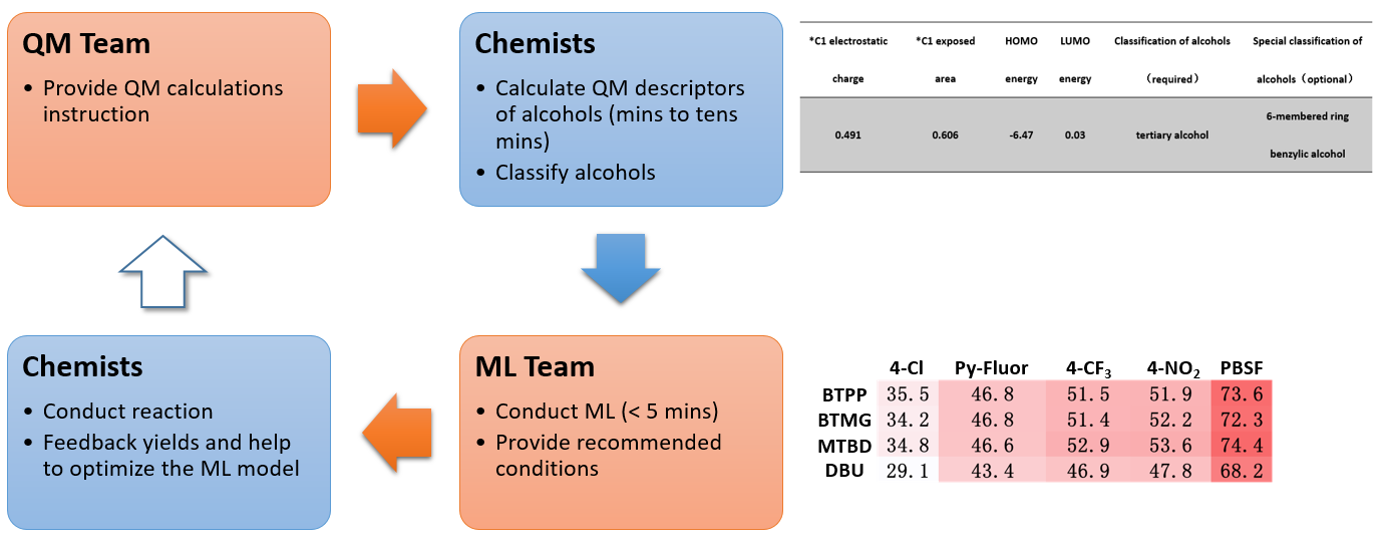
图7. 脱氧氟化反应预测服务流程
Doyle课题组报道了这么多成功的案例。如图8所示,我们在实际应用中,预测收率和分离收率是有相关性的。但也有一些例外。底物易消除,反应位点位阻大,还有临位基团参与效应,这些都会影响反应的收率。
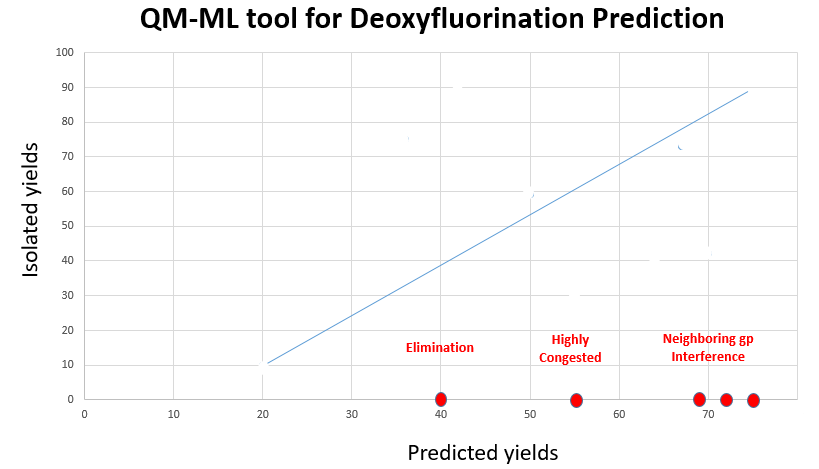
图8. 脱氧氟化预测收率和分离收率对比图
对于高活性易消除的醇和三级醇,以上的QM-ML模型并不能预测出合适的脱氧氟化条件,所以这里我们也推荐几个试剂供大家参考:高活性易消除的醇: SulfoxFluor[7]; 三级醇: Fluolead[8]和XtalFluor-E[9]。如果大家有什么好的试剂,敬请与我们分享。
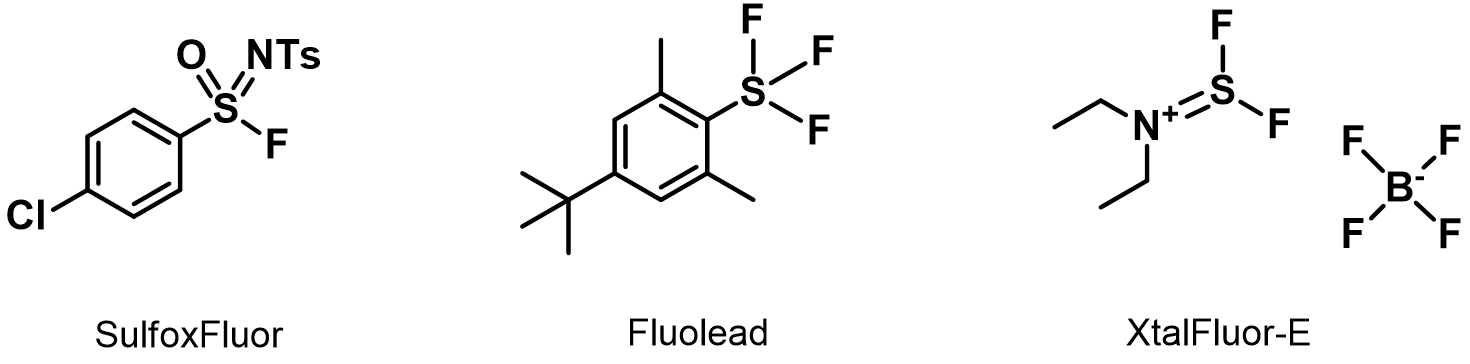
图9. 推荐几个有用的脱氧氟化试剂
本文由刘文锋、余晨、王浩薇、潘东、赖光华、卫小文编撰。
参考文献:
[1] D.E. Yerien, S. Bonesi, A. Postigo, Org. Biomol. Chem. 2016, 14, 8398.
[2] M.K. Nielsen, C.R. Ugaz, W. Li, A.G. Doyle, J. Am. Chem. Soc. 2015, 137, 9571.
[3] N.W. Goldberg, X. Shen, J. Li, T. Ritter, Org. Lett. 2016, 18, 6102.
[4] a. M.K. Nielsen, D.T. Ahneman, O. Riera, A.G. Doyle, J. Am. Chem. Soc. 2018, 140, 5004. b. The three substituted (Cl, CF3, and NO2) phenylsulfonyl fluorides are para substituted instead of meta substituted as described on p 5004 of this paper. They were prepared from corresponding commercially available para sulfonyl chloride. See paper’s supporting information S-21.
[5] F. Strieth-Kalthoff, F. Sandfort, M.H.S. Segler, F. Glorius, Chem. Soc. Rev. 2020, 49,6154.
[6] J.K. Guo, C.W. Kuang, J. Rong, L.C. Li, C.F. Ni, J.B. Hu, Chem. Eur. J. 2019, 25, 7259.
[7] T. Umemoto, R.P. Singh, Y. Xu, N. Saito. J. Am. Chem. Soc.2010, 132, 18199.
[8] S. Kalidindi, A.S. Gangu, S. Kuppusamy, S. Sathasivam, V. Shekarappa, S. Murugan, S. Bondigela, M. Kandasamy, K. Ghanta, A. Vinodini, A. Shrikant, R. Ramachandran, W.P. Gallagher, N. Kopp, F. González-Bobes, M.D. Eastgate, R. Vaidyanathan, Org. Process Res. Dev. 2021, 25, 1556.