 选择语言
选择语言



新闻和活动

关于我们
服务和解决方案
新闻和活动
职业发展
服务平台
QM有机化学课堂
市场活动
新闻
QM有机化学课堂
在前面的第七章和第十章[1]中,我们探讨了如何利用QM工具预测多卤代芳香底物亲核取代反应的反应位点及反应优先顺序。今天,我们将再给大家分享一个应用实例。
图1所示的2,3,6-三氟-4-溴苯甲醛与甲醇钠的亲核取代反应,苯环上有四个卤素,那么哪个位点的卤素活性最高呢?根据我们学到的有机合成知识,芳香亲核取代反应中氟的活性高于溴[2],醛基是吸电子取代基,所以2位和6位氟的反应活性较高。但我们却无法进一步判断反应会选择性的得到2位或6位取代的产物,还是会得到一对混合物。实际的反应结果是仅得到了2位氟原子被取代的产物,而没有分离到6位氟被取代的产物。为什么反应会有这么高的选择性呢?接下来,我们将尝试利用QM来解答这个问题。

图1. 2,3,6-三氟-4-溴苯甲醛与甲醇钠的亲核取代反应

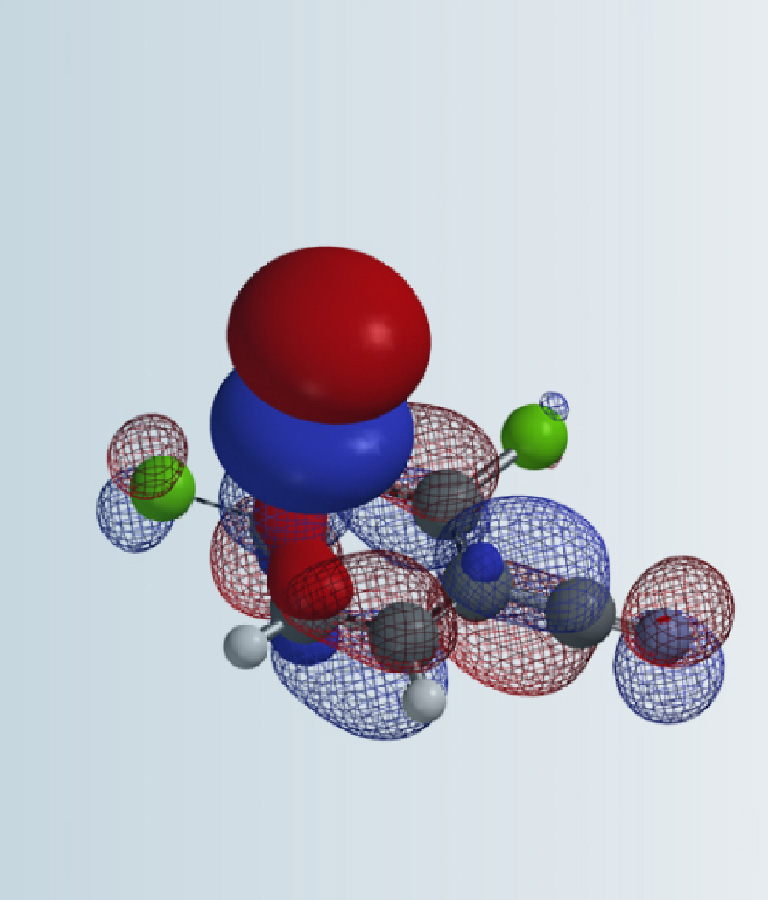
图2. 2,3,6-三氟-4-溴苯甲醛的LUMO、LUMO+1及LUMO/LUMO+1/Density图
首先计算得到底物的LUMO及LUMO+1,如图2所示。LUMO与LUMO+1的能量差较大 (DE = 1.66 eV),所以这里我们只需分析底物的LUMO即可。C2和C6 的LUMO lobe大小相似,很难判断哪个碳原子更容易被进攻。当我们进一步结合电子密度等值面[3]来分析,可以清晰地观察到LUMO lobe在2位比6位更易接近,意味着在相同条件下,2位被进攻的可能性更高。
接下来,我们分别对2和6位发生亲核进攻生成Meisenheimer络合物反应过程的能量变化曲线进行了计算。
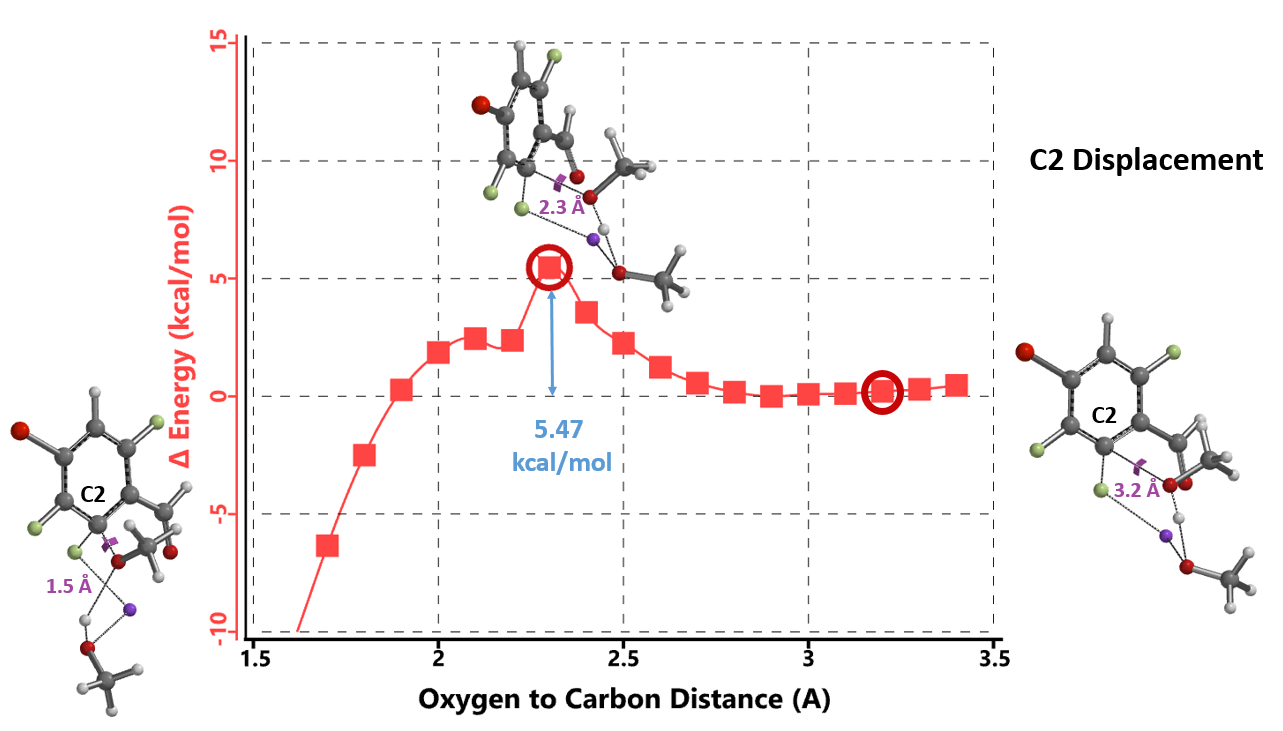
图3. NaOMe/MeOH与底物形成中间态的C-2和C-6活化能比较图
结果如图3所示,6位发生反应所需的活化能比2位要高出4.42 kcal/mol,反应更倾向于发生在2位,这与实验结果是一致的。由2位取代反应的能量变化曲线可知该反应所需的活化能相对较低,为5.47 kcal/mol,该反应应该在室温或更低的温度下就可以快速进行,但这显然与实际反应所需的实验条件(65度,12小时)不符。这是为什么呢?
复盘底物的LUMO (图2),我们发现醛基碳较C2和C6上有更大的LUMO lobe,其发生亲核进攻反应的活性应该更高。实际的反应历程可能和我们前面假设的过于简化的反应机理不同,所以导致QM预测的结果和实际实验条件存在较大的偏差。
我们认为反应过程中醛基可能首先与甲醇钠反应生成甲缩醛钠盐,然后才会发生芳香亲核取代反应。为了检验这个假设,我们计算第一步生成半缩醛钠盐的能量变化曲线(图4),发现这是一个能量下降的过程,没有任何能垒,说明参与SNAr反应的实际底物是半缩醛钠盐。

图4. 半缩醛钠盐生成反应的能量变化曲线
接下来设置半缩醛钠盐与甲醇钠的亲核取代反应进行计算,我们需要考虑有几个甲醇分子参与反应。如图5所示,根据底物结构特点,我们假设有一分子或两分子甲醇参与了反应,分别计算了其对应的能量变化曲线及过渡态结构。

图5. 一分子和两分子甲醇参与第二步反应的中间态的活化能比较图
通过计算得知,当有一分子甲醇参与反应时,6位发生反应所需活化能比2位高出1.40 kcal/mol (图5,左图);而有两分子甲醇参与第二步反应时,6位发生反应所需能量比2位高出2.19 kcal/mol (图5,右图)。二者的计算结果均倾向于2位更容易发生反应。使用两分子甲醇模型计算的反应能量变化曲线在 2.0 到 1.5 Å 之间表现出独特的能量平缓下降的平台2c(图 6,2位碳原子与亲核试剂氧原子的距离),对应于 21.45 kcal/mol 的能垒,与实验条件更一致 (65 ºC,12 小时)。这与实验结果是一致的。
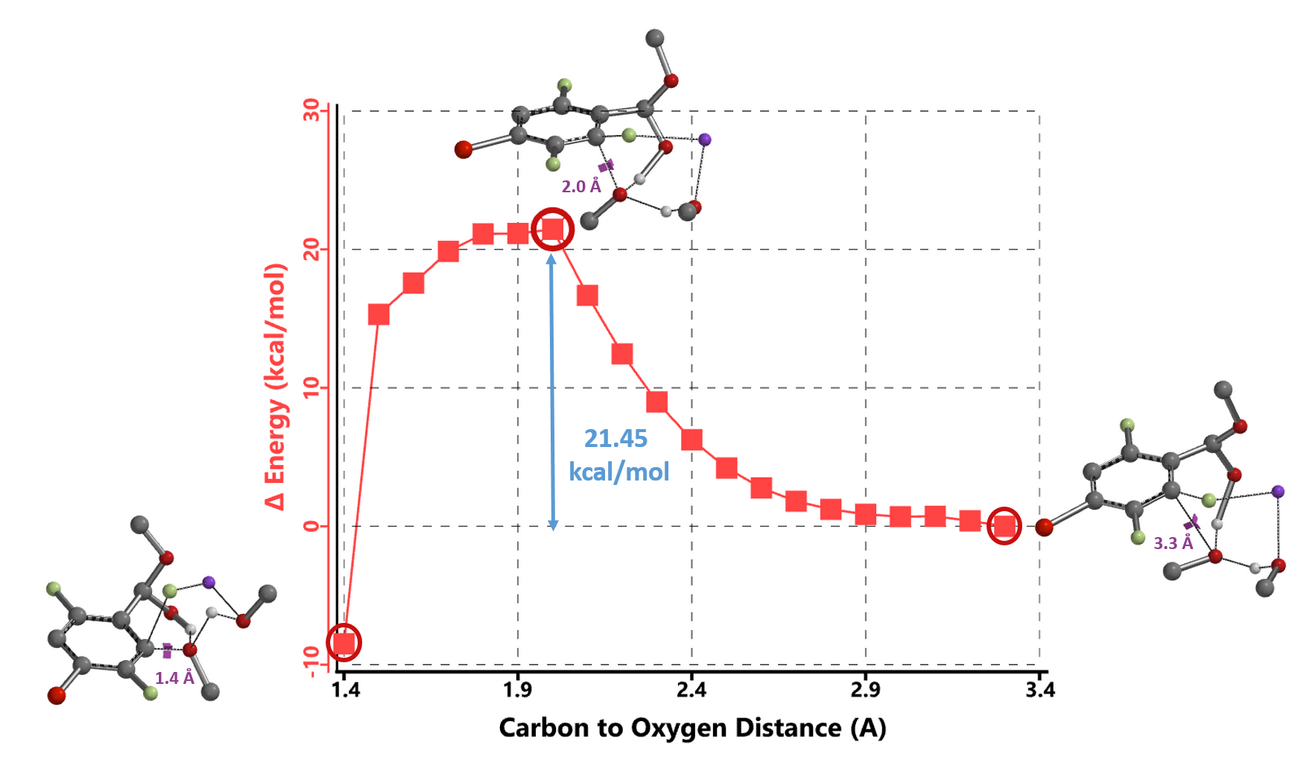
图6. 半缩醛钠盐与甲醇钠的亲核取代反应 (两分子甲醇反应模型) 的能量变化曲线
过渡态(iFreq i350 cm-1)和内禀反应坐标(IRC)计算都支持我们假设的半缩醛钠盐-两分子甲醇反应模型。我们认为在水相后处理时,SNAr 产物的半缩醛将水解回醛基(如图7所示)。
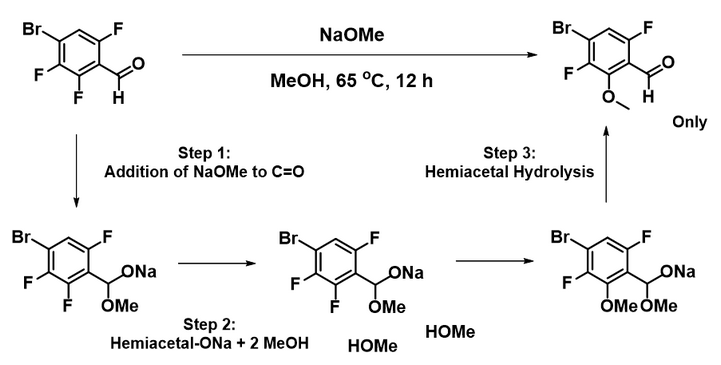
图7. 2,3,6-三氟-4-溴苯甲醛与甲醇钠半缩醛机制的SNAr反应历程的化学式展示
总结一下,QM 计算2,3,6-三氟-4-溴苯甲醛和甲醇钠之间的 SNAr 反应是通过其相应的半缩醛产物进行的,溶剂甲醇分子是反应过渡态中不可或缺的组成部分。 一分子和两分子甲醇反应模型都可以解释 C2 优先发生反应的原因。使用两分子甲醇模型计算得到的反应活化能为 21.45 kcal/mol,与实验条件更吻合。
对于图 8 中的2-氟吡啶类的 SNAr 反应,间位有吸电子基团的(例如含CHO、CN 等)被认为会增加C-F碳的亲电性,从而加速SNAr反应。但在实践中,它们的反应活性非常低,不到5%的转化率。而当R为溴时,底物却很容易与仲胺发生SNAr,收率高达80%。根据LUMO、LUMO+1 及其与电子密度图的叠加图,您能给出合理的解释吗?
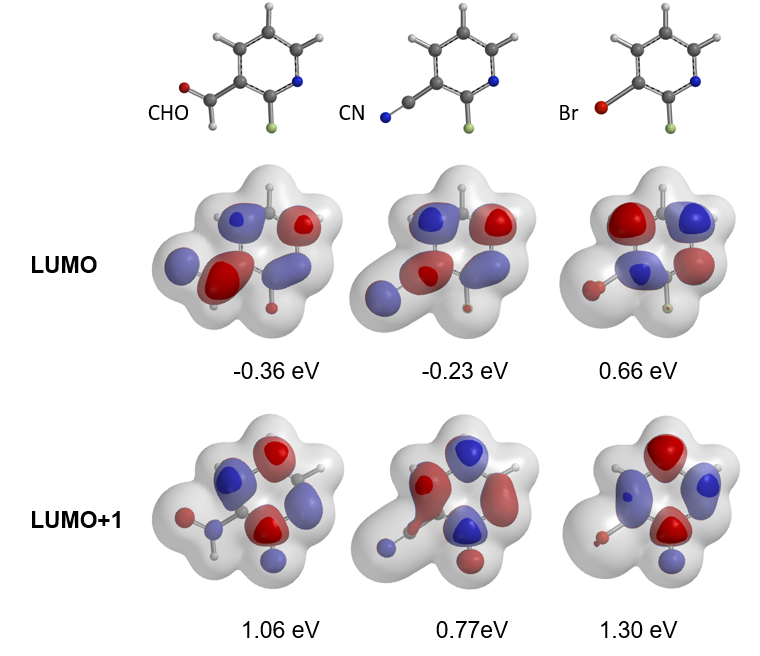
图8. 2-氟吡啶与仲胺的反应(上图);底物LUMO、LUMO+1与电子密度图叠加图(下图)
本文由谭大金、陈永胜、董立亭、卫小文编撰。
参考文献:
[1] QM有机化学课堂第七章《LUMO运用进阶如何预测亲核反应选择性》、第十章《QM在多卤素芳香底物亲核反应中的应用》。
[2] (a) S. Rohrbach, A.J. Smith, J.H. Pang, D.L. Poole, T. Tuttle, S. Chiba S, J.A. Murphy, Angew. Chem. Int. Ed., 2019, 58, 16368. (b) O. Acevedo, W.L. Jorgensen, Org. Lett., 2004, 6, 2881. (c) E.E. Kwan, Y. Zeng, H.A. Besser, E.N. Jacobsen, Nature Chemistry, 2018, 10, 917.
[3] Spartan’20 Tutorial and User’s Guide (2020). Irvine, CA, USA: Wavefunction, Inc. pp362-368.