 选择语言
选择语言



新闻和活动

关于我们
服务和解决方案
新闻和活动
职业发展
服务平台
QM有机化学课堂
市场活动
新闻
QM有机化学课堂
通过半胱氨酸残基的巯基衍生化蛋白质是一种流行的生物偶联方法,如图1所示,巯基衍生化的高活性官能团有碘乙酰胺、马来酰亚胺和二硫键等。其中,碘代乙酰胺官能团已经在蛋白质标记中得到了广泛的应用,近年来的研究表明氯代乙酰胺比碘代乙酰胺在半胱氨酸残基衍生化蛋白质过程中表现出更好的选择性[1]。在第二十六章我们初步讨论了由于氢键在烷基化反应中的作用,反应得到异于经验的位置异构产物的机理;在文末的“小试牛刀”部分,提到硫醇与N-甲基氯乙酰胺和N,N-二甲基氯乙酰胺的亲核取代反应,在速率上存在明显差异。本章节就以上两种试剂与硫醇的亲核取代反应进行QM考察分析。

图1. 用于构建高DAR值的生物偶联物策略示例[2]
从图2可以看出,N-甲基氯乙酰胺和N,N-二甲基氯乙酰胺作为亲电试剂,与半胱氨酸残基发生烷基化反应时,前者的反应速率会明显慢于后者。这和我们的认知相悖,因为N,N-二甲基氯乙酰胺的位阻更大,理应反应更慢。是什么原因造成如此大的反应速率差距呢?我们首先考察了N-甲基氯乙酰胺和N,N-二甲基氯乙酰胺的LUMO和LUMO map,如图3。
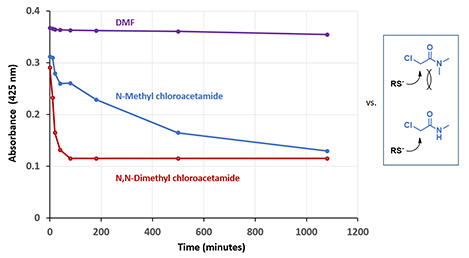
图2. N-甲基氯乙酰胺和N,N-二甲基氯乙酰胺作为亲电试剂的反应活性

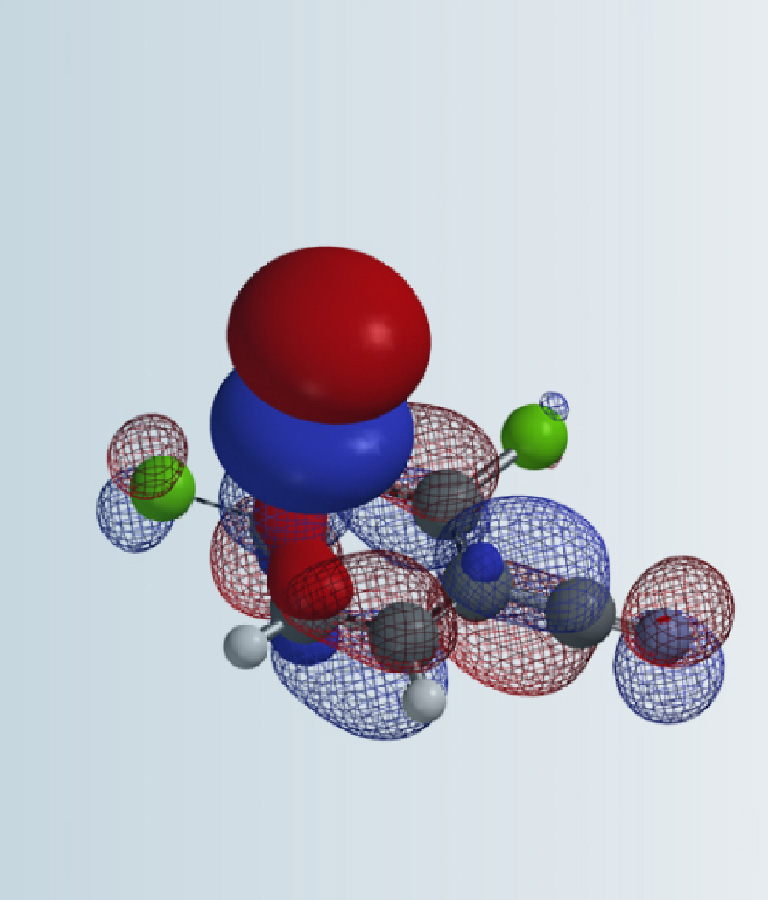
图3. N-甲基氯乙酰胺和N, N-二甲基氯乙酰胺的LUMO和LUMO map (IsoVal 0.002 e/au3, 99.2%)
N-甲基氯乙酰胺的LUMO值为2.64 eV, LUMO map和inaccessibility marker表明,亚甲基碳原子不易被接近。而N,N-二甲基氯乙酰胺的LUMO值较低,为1.74 eV,并且亚甲基碳原子的LUMO lobe暴露的更充分,更容易接受亲核试剂的进攻,这意味着N,N-二甲基氯乙酰胺的反应活性较高,与实验结果相吻合。两个底物的LUMO能量及LUMO Map所示的反应位点可接近性是反应差异性的原因之一。为了进一步考察两种亲电试剂发生烷基化反应的明显速率差异,我们继续计算烷基化反应能量曲线。
为了节省时间和资源,我们将亲核试剂简化为甲基硫醚负离子,分别考察N-甲基氯乙酰胺和N,N-二甲基氯乙酰胺发生亲核取代反应的活化能。我们首先计算N,N-二甲基氯乙酰胺和甲基硫醚负离子发生亲核取代反应的活化能,如图4所示。(计算级别为B3LYP-D3/6+31G*,下同)

图4. N,N-二甲基氯乙酰胺进行亲核取代反应的活化能曲线
由图4可知整个反应的活化能相当低,仅有约5.6 kcal/mol,并且活化能曲线在3.0埃和2.6埃时各有一个能量峰值。为了理清反应的具体过程,我们对取代反应中复合物的构象进行了考察,如图5。可以看到在硫原子距亚甲基碳3.5埃时,酰胺基团和氯原子基本共平面,由于硫负离子发生亲核取代反应需要从氯原子的背面进攻,这就意味着要克服二甲基的空间位阻。随着硫原子逐渐接近亚甲基碳,二甲胺基和氯原子共平面的趋势开始被破坏,直到距离为2.9埃时,整个酰胺基团旋转到和碳氯键垂直,而后硫原子发生亲核取代反应,该反应的能垒只有0.88 kcal/mol。
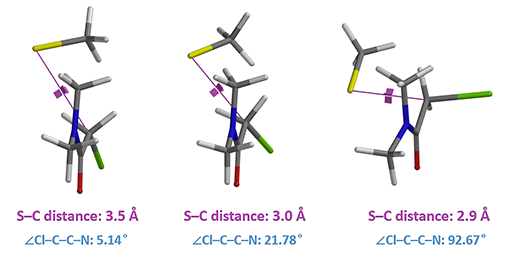
图5. N,N-二甲基氯乙酰胺进行亲核取代反应中的构象变化及∠Cl–C–C–N的二面角
随后我们计算得到N-甲基氯乙酰胺发生亲核取代反应的活化能曲线图,如图6所示。该反应所需活化能显著高于N,N-二甲基氯乙酰胺,达到了10 kcal/mol,且活化能是一个单峰的曲线。N-甲基氯乙酰胺的亲核取代过程中存在何种阻力,能让整个取代反应的活化能增加约5 kcal/mol,让我们继续通过考察反应历程中的构象变化来找出原因。

图6. N-甲基氯乙酰胺进行亲核取代反应的活化能曲线图
从图7中我们可以看到,硫原子距离亚甲基碳3.5埃时,氯原子和酰胺近似垂直,而硫原子、亚甲基碳和酰胺近似于共平面,硫原子和氢原子的距离只有约2.1埃,这要显著小于碳原子和硫原子的范德华半径之和(2.9埃)[3];另一方面,酰胺氮氢键也发生了明显的伸长(1.057-1.011埃),这意味着酰胺氢原子和硫原子之间存在氢键的相互作用。当硫原子离亚甲基碳3.0埃时,尽管氯原子和酰胺变为了共平面状态,但是从硫原子和酰胺氢原子的距离和二面角来看,相互作用仍然存在。硫原子进一步接近亚甲基碳时,氯原子和酰胺慢慢变为相互垂直,硫原子和酰胺脱离了共平面状态,随着酰胺氢原子和硫原子相互远离,酰胺氮氢键键长也收缩到近似标准键长。从复合物的电子云密度切面也可以看出(此切面处S,C,N共平面),在硫原子距离亚甲基碳较远时,硫原子和氢原子间存在一个电子云密度较高的区域,而这样一个高密度区域会随着硫原子接近亚甲基碳原子而逐渐消失,硫和酰胺键之间的氢键逐渐断裂,开始发生烷基化反应。

图7. N-甲基氯乙酰胺进行亲核取代反应中的构象变化及∠S–C–C–N的二面角
N-甲基氯乙酰胺的取代反应可以认为是按照图7中第二个空间结构进行的。这里甲基硫负离子并不需要克服N,N-二甲基氯乙酰胺例子中的位阻影响,但是它需要完全消除和酰胺氮氢的相互作用才能够到达反应的过渡态。从化合物构象上来说,即只有当碳氯键和酰胺键平面相垂直时,取代反应才能够发生。
从这个例子中我们可以看出,当反应复合物中存在氢键作用时,它既可以通过稳定反应的过渡态来加速反应的进行;另一方面,如果需要克服氢键作用才能到达反应的过渡态,那么此处氢键的存在就阻碍了反应的进行。如本例中存在的分子间氢键就成为了发生亲核取代反应的一个阻力。
考察不同底物的反应活性,有时只考虑反应物的空间位阻和电子效应是不够的,我们需要将分子间相互作用力也纳入考查范畴。在分子间作用力中,由于氢键的能量较为显著,可能对反应的过程产生显著的影响,故在探讨反应历程中需要特别注意。此外,QM计算工具让我们能够用客观的计算数据量化该类交互作用,合理的解释我们在实验中观察的现象,加深对反应的理解。
SNAr反应通常在极性非质子溶剂(DMSO, MeCN, DMF, etc)中比在极性质子溶剂中更容易得到好的效果。1-氟-4硝基苯和叠氮负离子的在不同溶剂中发生SNAr反应的结果如表1所示[4]:

表1. nBu4NN3与1-氟-4-硝基苯在25℃下不同溶剂中的反应速率常数(log k)[4]
在实验室工作中,我们会遇到一些特殊的SNAr反应。如伯胺与2-氟吡啶在DMSO、NMP、MeCN中完全不反应,但在醇中很容易进行。理论上来说,伯胺在极性质子性溶剂中高度溶剂化,它们在醇中的反应应当比在极性非质子溶剂中慢。但我们观察到完全相反的结果,原因为何?
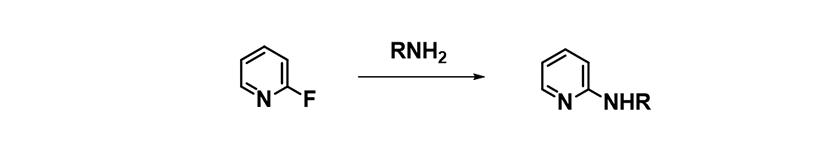
图8. 2-氟吡啶与伯胺的SNAr反应
本文由王守亮、郑重、王秋月、王健、卫小文编撰。
参考文献:
[1] https://www.researchsquare.com/article/rs-356231/v1
[2] J. Kalia, R. T. Raines, Curr. Org. Chem. 2010, 14, 138.
[3] P. Atkins, J. de Paula, J, Keeler, Atkins’ Physical Chemistry (11th ed.). Oxford University Press. Oxford, 2018: pp 584
[4] B.G. Cox, A.J. Parker, J. Am. Chem. Soc. 1973, 95, 408.