 选择语言
选择语言



新闻和活动

关于我们
服务和解决方案
新闻和活动
职业发展
服务平台
QM有机化学课堂
市场活动
新闻
QM有机化学课堂
在有机合成中,有一个问题大家一定遇到过:相同类型的反应,有些底物可以顺利发生,有些收率很低,有些则很难进行。以我们最常见的卤代反应为例,富电子的芳环可以在氯代丁二酰亚胺 (NCS) 条件下顺利氯化,缺电子底物的卤代反应却难以发生,这时候就需要尝试二氯海因或者三氯异氰脲酸。经验告诉我们二氯海因或者三氯异氰脲酸比NCS有更强的氯代反应活性,但是有多强呢,有没有一个具体的数值来衡量呢?

图1. 三氯异氰脲酸、二氯海因和NCS的LUMO轨道及能量
如图1所示,分别计算三氯异氰脲酸、二氯海因和NCS的LUMO轨道和能量。这些试剂的LUMO lobes均分布在氯原子上,而不是在C=O碳上,所以他们是卤化试剂,而不是酰化试剂。三氯异氰脲酸的LUMO能量为-0.79 eV,二氯海因为0.37 eV,NCS为1.09 eV。活性最高的三氯异氰脲酸LUMO能量最低,活性相对较低的二氯海因LUMO能量增加,活性最低的NCS有最高的LUMO能量。根据这三个试剂的计算结果和实验观察到的反应活性,我们可以总结出一个大概的规律:LUMO能量越低,反应活性也就越强,LUMO能量可以用来衡量卤化试剂的活性。
利用HOMO和LUMO的能量差评估反应活性
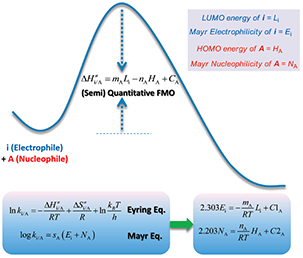
图2. 前线分子轨道理论理解Mayr方程
为什么LUMO能量可以衡量反应活性呢?余志祥小组给出了其中的理论解答[1]。如图2所示,他们从前线轨道理论 (FMO) 出发,结合过渡态理论中的Eyring方程,推导出Mayr方程和FMO理论在原理上是一致的。当在比较一系列分子的反应活性时,如果影响这些分子的反应性主要是由于不同取代基的电子效应所引起的,那么,一个分子的亲核性和该分子的最高占据轨道HOMO相关,而亲电性则与该分子的最低空轨道LUMO相关。也就是说可以通过HOMO能量和LUMO能量评估底物的亲核性和亲电性强弱,进一步通过HOMO和LUMO的能量差来判断反应活性。
接下来,我们就应用LUMO-HOMO能量差的方法来解释分子内Pictet-Spengler的反应趋势。
Pictet-Spengler反应是β-芳基乙胺或者β-杂芳基乙胺与醛或酮在酸性条件下发生缩合,之后形成的亚胺和芳环发生亲电芳香取代得到关环产物 (图3,上)。该反应的驱动力是醛和胺缩合生成亚胺的亲电性和芳基/杂芳基部分的亲核性之间的相互作用。

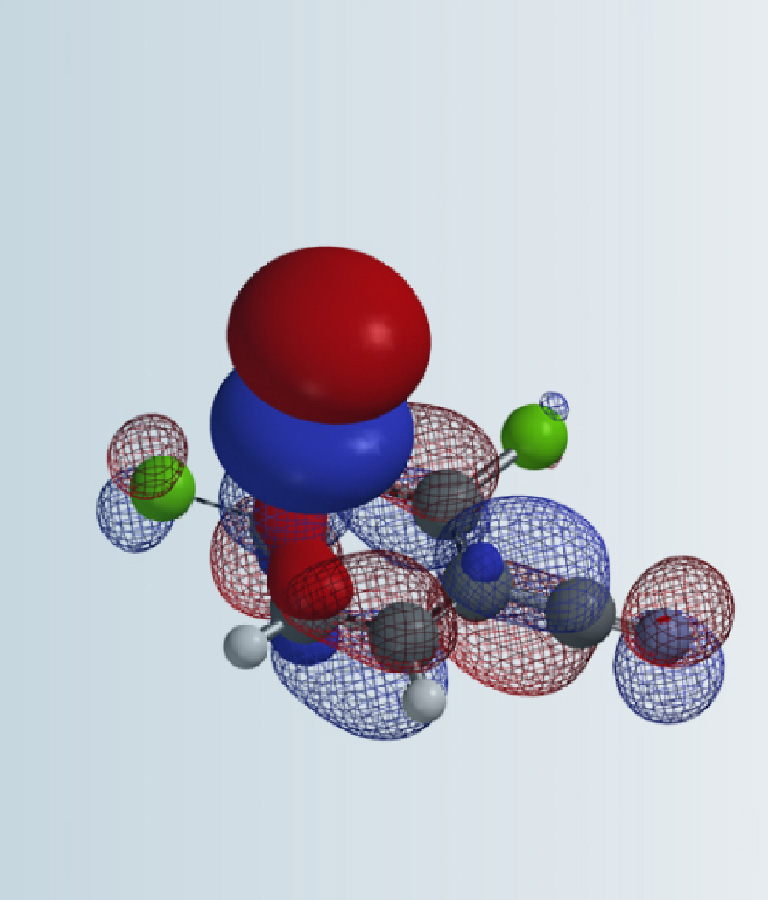
图3. Pictet-Spengler 反应(上)和亚胺的LUMO轨道、HOMO轨道及能量(下)
如图3所示,从亚胺化合物的HOMO图上,可以清楚地看到吲哚C2原子上的HOMO lobes分布。从LUMO图上,也可以看到亚胺碳上的LUMO lobes分布。所以该分子的Pictet-Spengler反应活性可以用LUMO和HOMO轨道的能量差来表示,两者的差值为8.1 eV。如果反应位点没有HOMO lobes 和LUMO lobes分布,就需要考虑有lobes分布的HOMO-1和HOMO-2或者LUMO+1和LUMO+2等来参与能量差计算。
接下来保持亚胺部分不变,将吲哚环替换为其它芳环,并计算一系列亚胺中间体的LUMO和HOMO轨道的能量差 (计算数据见表1,结构见图4)。
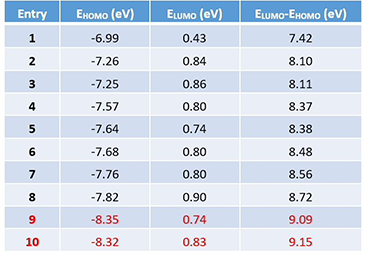
表1. 不同骨架Pictet-Spengler反应亚胺中间体的LUMO和HOMO轨道能
如图4所示,随着LUMO和HOMO之间的能量差逐渐增大,需要更高的温度或者更强的酸才能发生环化反应[2]。当能量差超过约9 eV的阈值时,则无法让反应进行。
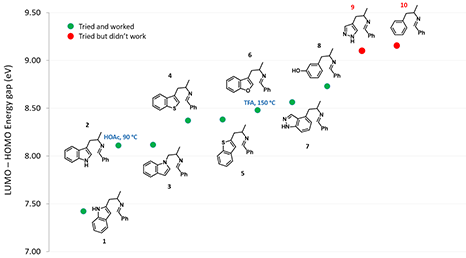
图4. LUMO-HOMO 能量差解释不同底物发生Pictet-Spengler反应的难易程度
在以上计算的基础上,我们就能以化合物9吡唑杂环化合物的能量差值9.09 eV为界限,来预测某一个化合物是否能够发生Pictet-Spengler反应。
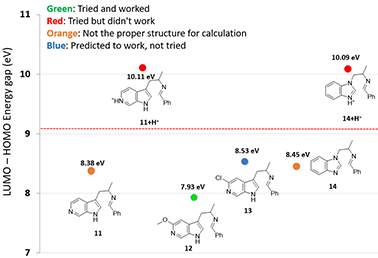
图5. 利用LUMO-HOMO 能量差预测不同底物的反应活性 (11+H+能量差由LUMO+1和HOMO-2计算,14 能量差由LUMO和HOMO-1计算,14+H+能量差由LUMO+2和HOMO-3计算)
对于含碱性氮的底物,我们在考虑能量差的同时,还要考虑底物的pKa,确定亚胺中间体是否质子化。
如图5所示,6-氮杂吲哚底物11,没有质子化时,LUMO-HOMO能量差为8.38 eV,处在可以发生反应的范围。但是,实际反应并不能发生,文献检索也找不到任何成功案例。这是因为在酸性反应体系中,吡啶氮原子质子化 (吡啶质子化pKa约为5.2),11+H+的LUMO-HOMO能量差高达10.11 eV。
团队想到一个巧妙的方法来解决这个问题:在N的邻位引入一个取代基,甲氧基或氯,来降低N原子的碱性 (2-MeO-和 2-Cl 吡啶的pKa分别是 3.2 和 0.7)[3],之后将取代基脱除。计算MeO取代底物12和Cl取代底物13的LUMO-HOMO能量差分别是7.93 eV和8.53 eV,均处在可以发生反应的范围内。我们决定首先尝试合成MeO产物,因为12的LUMO-HOMO能量差7.93 eV比9.09 eV的界限远多一点。实际合成中亦顺利得到了甲氧基取代的反应产物,随后将OMe转化为OTf,顺利脱除OTf得到6-氮杂吲哚产物[4]。
通过提前计算,苯并咪唑类底物14也会出现类似的问题,质子化14+H+的能量差高达10.09 eV,这就警示我们此路不通,要寻找其他方法啦[4]。
本章向大家介绍了利用LUMO和HOMO的能量差快速评估反应活性的方法。相对于计算过渡态,这种方法更加简便快捷,对于一些已经有大量反应实例的基础反应尤其适用。通过总结反应底物的HOMO和LUMO能量范围,就可以评估出新底物的反应活性。但是需要注意的是,这种方法仅适用于电子效应是反应活性主要影响因素的情况,如果有明显的空间效应则不适用。此外,还需要注意根据反应位点的轨道lobes可用性选择正确的轨道,可能会是HOMO-1和HOMO-2,LUMO+1和LUMO+2来计算相关能量。
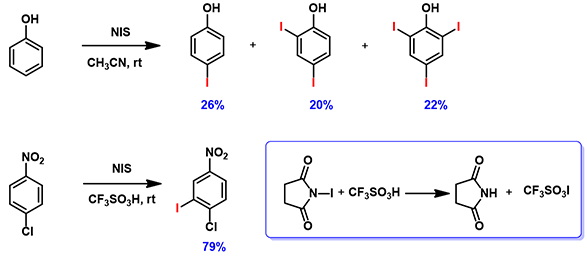
图6. 苯酚和4-氯硝基苯的碘代反应
如图6所示,在NIS的卤化反应中,苯酚可以得到双取代和三取代的产物[5]。4-氯硝基苯在同样条件下则难以发生卤化反应,但是加入三氟甲磺酸后却以79%的收率得到产物[6]。文章认为三氟甲磺酸与NIS反应生成三氟甲磺酸碘,是反应中真正的卤化试剂。利用本章的知识来解释一下吧!

图7. NIS和CF3SO3I的LUMO轨道以及苯酚和4-氯硝基苯的HOMO轨道及相应的能量
你会选择什么试剂去碘化1,2,3-三氟-4-硝基苯呢?卤化会发生在C-5位还是C-6位呢?
开动大脑,留言反馈,神秘大奖等着你!
参考文献:
[1] L.G. Zhuo, W. Liao, Z.X. Yu, Asian J. Org. Chem. 2012, 1, 336.
[2] A. Yokoyama, T. Ohwada, K. Shudo, J. Org. Chem. 1999, 64, 611.
[3] https://www.chem.wisc.edu/areas/reich/pkatable/pKa_compilation-1-Williams.pdf
[4] M.K. Jackl, I. Kreituss, J.W. Bode, Org. Lett. 2016, 18, 1713.
[5] P. Bovonsombat, J. Leykajarakul, C. Khan, K. Pla-on, M.M. Krause, P. Khanthapura, R. Ali, N. Doowa, Tetrahedron Lett. 2009, 50, 2664.
[6] G.A. Olah, Q. Wang, G. Sandford, G.K. Surya Prakash, J. Org. Chem, 1993, 58, 3194.