 选择语言
选择语言



新闻和活动

关于我们
服务和解决方案
新闻和活动
职业发展
服务平台
QM有机化学课堂
市场活动
新闻
QM有机化学课堂
1,2,3-三氮唑是药物化学中的常见骨架,具有该结构的化合物往往呈现广泛的生物活性。1,4-取代的 1,2,3-三氮唑作为基本的药效团,具有多功能性(图 1):它可以作为特定功能基团的生物等效替代物;作为分子支架,它调节其他药效基团维持活性构象;可以作为药效基团原件的链接单元;基于其对蛋白酶的稳定性,发挥拟肽作用;在多种情况下作为连接子发挥作用,比如作为 PROTACs 的连接子;药物载体的连接子;靶向结合物或者探针的连接子等等[1]。
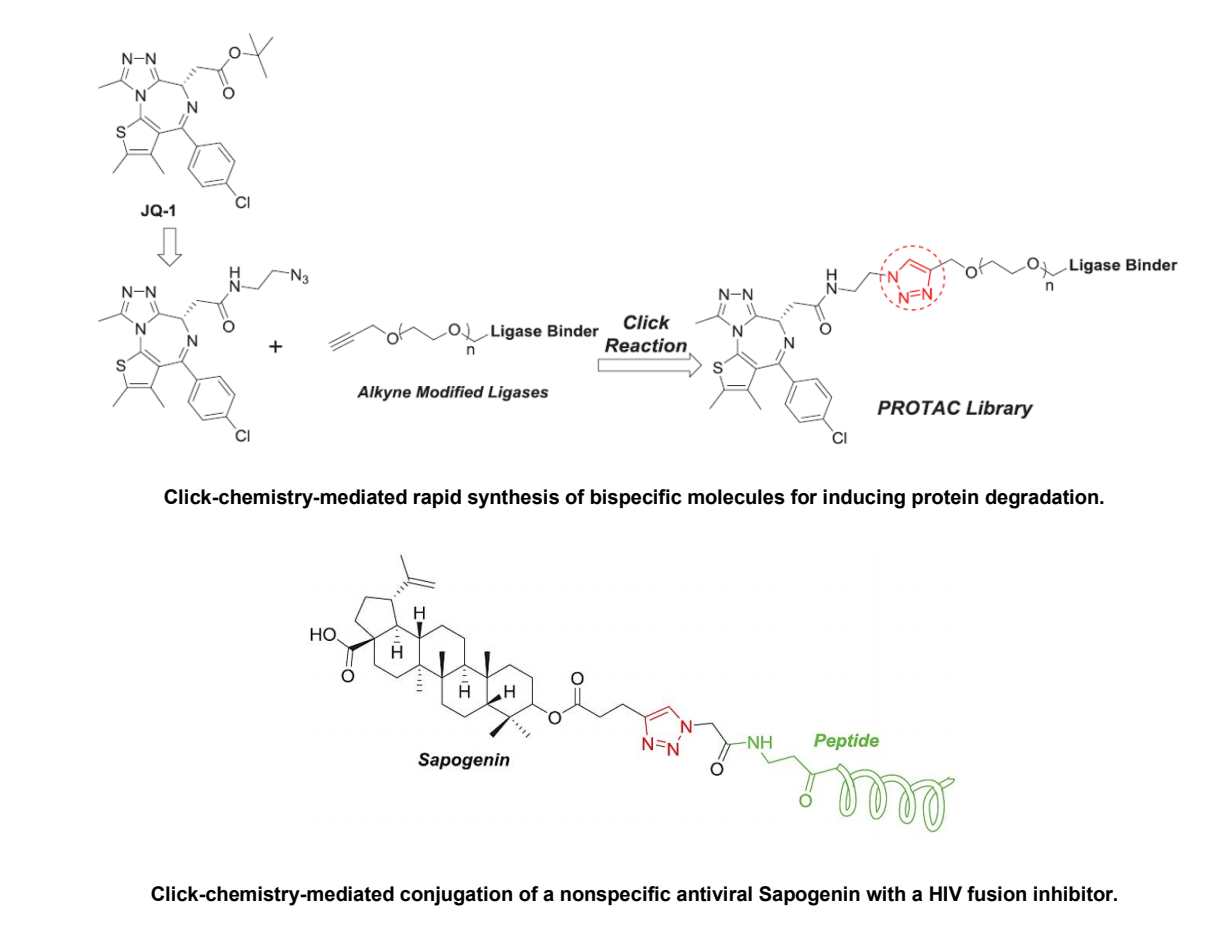
图 1. 1,2,3-三氮唑骨架在药物合成中应用示例
1,2,3-三氮唑结构一般通过传统的[3+2]环加成反应,以及近些年由 Sharpless 教授提出的Click chemistry(图 2)构建。两种方法都需要叠氮化合物作为起始原料,而叠氮基为高活性官能团,常具有剧毒和易制爆的特性,因此,安全制备和使用叠氮化合物至关重要。
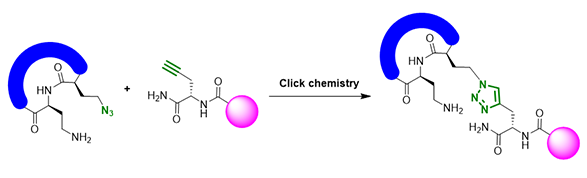
图 2. 1,2,3-三氮唑骨架构建示例
2019 年,中科院上海有机所的董佳家教授和 Sharpless 教授在 Nature 上发表了一种安全高效制备各种叠氮化合物的全新方法,并将其用于模块化 Click 反应,构建含上千个分子的化合物库(图 3)[2]。

图 3. 发表在 Nature 上的重氮转移反应
文中介绍的磺酰氟类化合物 FSO2N3(氟磺酰基叠氮),表现出对于一级胺官能团异乎寻常的重氮转移反应性。相对于传统的重氮转移反应,该反应呈现诸多优点,如室温即可以进行,反应底物是化学计量(1:1)的,短时高效转化,方法适用于所有的一级胺,包括叔丁基伯胺都可以实现接近化学计量转化。本节 QM 课堂,我们就对该重氮转移反应进行学习探讨。
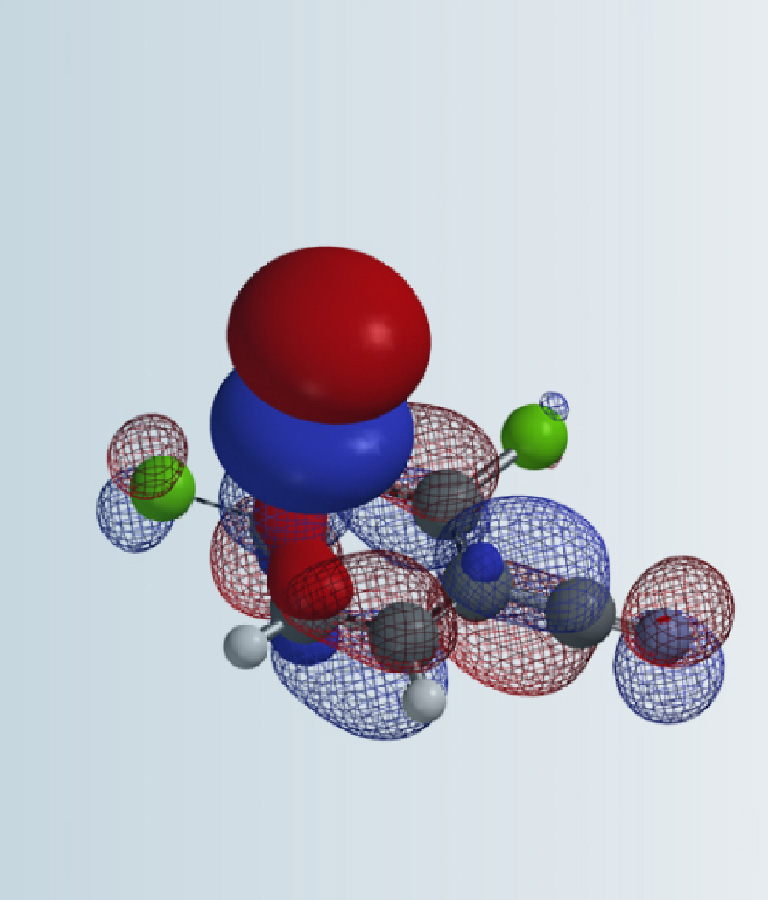
我们知道,叠氮基团是两性基团,其亲电性使它与胺作为亲核试剂的重氮转移反应成为可能。首先,我们需要判断胺基的亲核进攻发生在叠氮的哪个氮原子上(图 4)。

图 4. 磺酰氟叠氮 LUMO 示意图
首先计算磺酰氟叠氮的 LUMO 和 LUMO map,结果如图 5 所示。可以看到三个 N 原子的LUMO lobe 大小顺序 N-3 > N-2 > N-1,而 LUMO map 清晰的显示出 N-3 比 N-2 更容易被接近,因此胺的亲核进攻更倾向于发生在N-3上。这与2014年发表在Tetrahedron Lett. 的“Mechanistic studies on the diazo transfer reaction”文章阐述的 15N 同位素标记实验的结果一致[3]。

图 5. 磺酰氟叠氮的 LUMO 和 LUMO map 示意图
下面我们开始建立这个反应的计算模型。文章中的实验结果表明,水在重氮转移反应中起到了关键的作用。在前期大量 QM 计算的基础上,我们认为胺基对叠氮加成是一个分步机理,而非协同机理(图 6)。首先胺进攻磺酰氟叠氮的 N-3,接下来通过一个水分子参与的七元环过渡态,将一个氢原子转移到磺酰氟叠氮的 N-1 原子上。

图 6. 胺基对叠氮加成的可能机理
我们首先建立胺基进攻步骤的 QM 计算模型。经过前期计算分析,我们发现在这样的一个模型中,水的参与至关重要。胺的氮原子直接进攻磺酰氟叠氮的 N-3 原子,在没有水参与的情况下活化能大于 20 kcal/mol。而在这个模型中加入一分子水,通过分子间氢键作用来同时稳定叠氮和胺,将 N-N 键的距离变化设定为从彼此间无相互作用的 2.4 Å 到足以形成共价键的距离1.5 Å,步长 0.1 Å。计算结果表明水分子的参与能够大大降低该步的活化能,其能垒只有不足5 kcal/mol(图 7-1)。

图 7-1. 经由水分子辅助的胺基进攻叠氮 N-3 原子的能量变化曲线
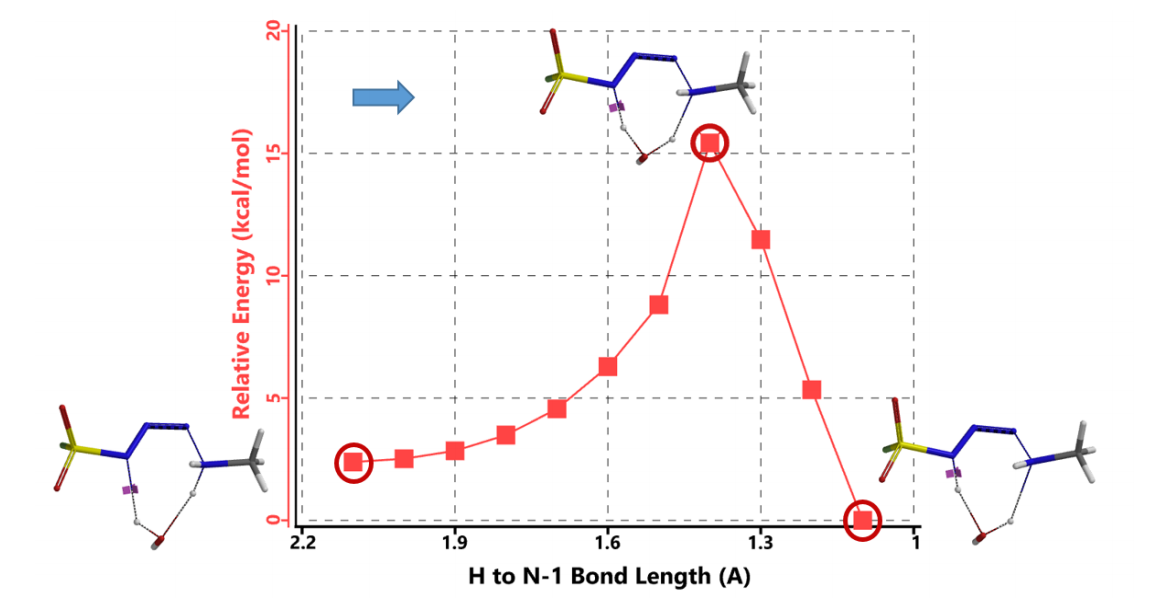
图 7-2. 经由水分子辅助的氢原子转移的能量变化曲线
接下来,我们以 N-N 键成键后,最低能量的结构作为接下来氢原子转移步骤的起点,计算这一步的活化能。结果表明该步的活化能也只有约 15 kcal/mol,在室温下同样能够顺利发生(图 7-2)[4]。
我们分别选取两个过程中能量最高点的结构,选择 Transition State Geometry 计算更加精确的过渡态能量,同时通过频率分析计算过渡态的虚频。结果表明两个过渡态结构均有且只有一个虚频,分别为 i136 cm-1 和 i1280 cm-1,见图 8。虚频相对于红外波谱中能观测到的各种分子振动的频率,并不是一个真实存在的频率。我们可以将其看做反应过渡态的结构,在向其能量最低状态运动的趋势,而将这个趋势近似为简谐运动时,所得到的频率即为虚频。当虚频只有一个时,我们可以认为在该点上的运动趋势仅有一组,即分别向着两个能量最低的状态运动(起始物和生成物),这意味着计算出的过渡态是合理的。
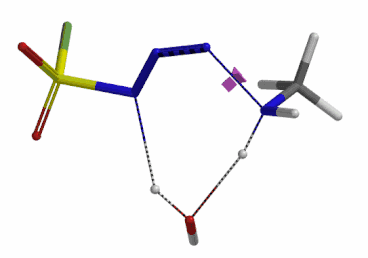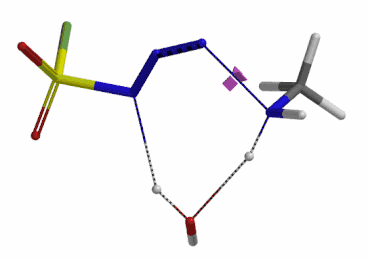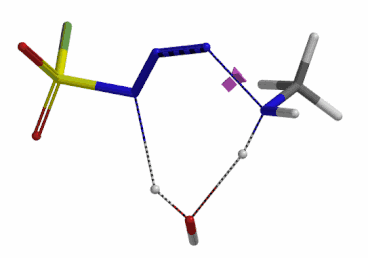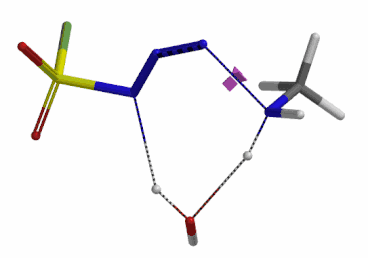
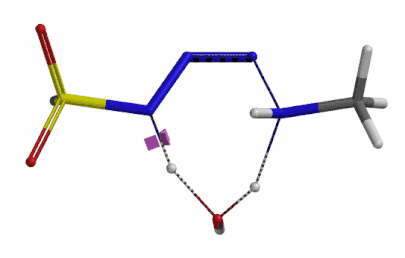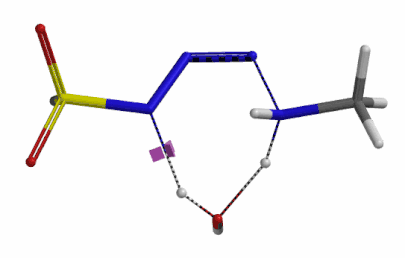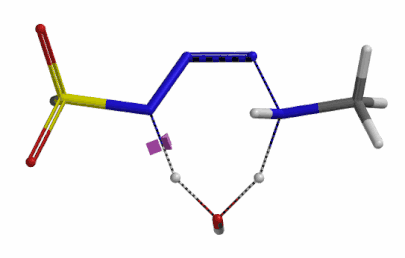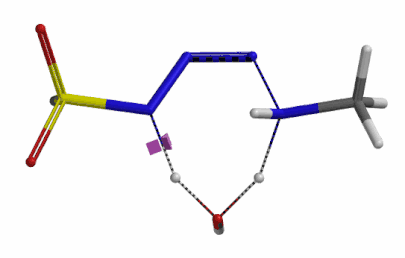
图 8. 胺基进攻叠氮 N-3 原子和氢原子转移步骤的过渡态结构及虚频136 cm-1 (左) 1280 cm-1 (右)
我们接下来计算第二步,即磺酰氟叠氮的 N-N 键断裂,以及第二个氢原子通过水分子,转移到磺酰氟叠氮 N-1 原子上的过程。经过计算,在没有水分子参与的时候,叠氮基团无法捕获到氢原子,导致反应无法发生。而在水分子参与其中时,这样一个过程的活化能约为 20 kcal/mol。这一步具有整个反应历程中最高的能垒,说明这一步是整个反应的决速步(Rate-Limiting Step)。对过渡态结构进行频率分析同样只得到了一个虚频,为 i239 cm-1,证实了这样一个过渡态的合理性(图 9)。
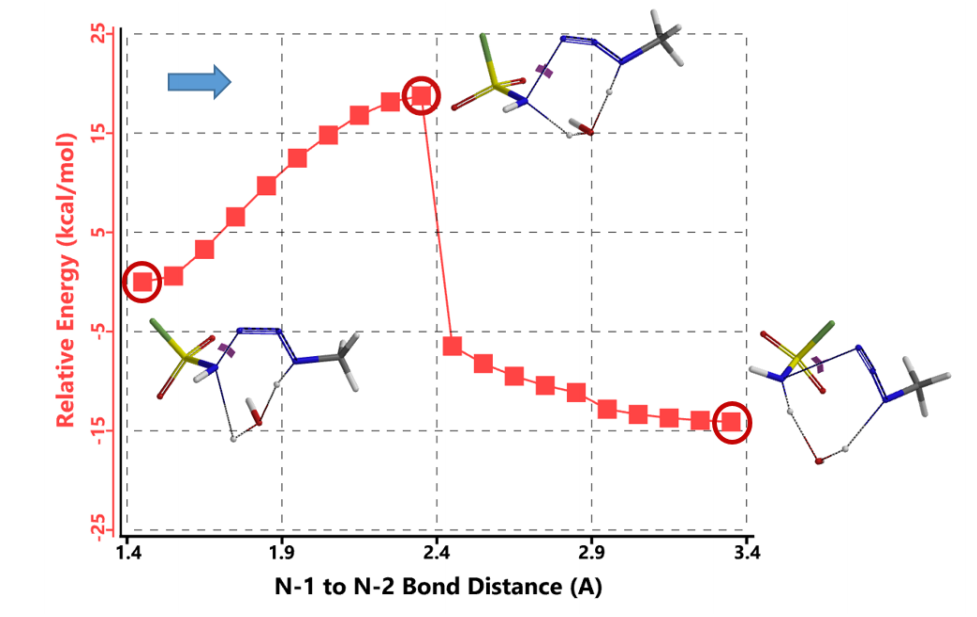
图 9. 磺酰氟叠氮 N-N 键断裂,氢原子转移并生成终产物的势能变化曲线
我们将起始物种的相对能量设为零,得到的反应中间体及过渡态的能量绘制在一张图中,即得到了这个重氮转移反应的势能剖面图,如图 10 所示。我们可以看到,反应的各个分步的能垒都很低,产物具有最低的能量,并且最后一个步骤的逆向过程会有约有 30 kcal/mol 的能垒,这样的差值意味着整个反应进程在室温下是不可逆的[4]。

图 10. 重氮转移反应的势能剖面图
这个例子展示了一个通过 QM 计算来推测反应历程的完整过程。首先,我们根据经验得出可能的机理和分步反应的第一个过渡态,并通过活化能计算得到该步反应的势能变化曲线,并通过频率分析的手段,验算虚频对过渡态的合理性进行进一步的确认。接下来我们以该过渡态产物的构型出发,重复上述这一过程,直到反应的过渡态能够导向最终产物,同时也再一次指出水在反应中的重要作用。这为我们系统的研究反应机理提供了一个得力的手段,希望大家能从这个例子中得到借鉴,并应用到自己的工作中去。
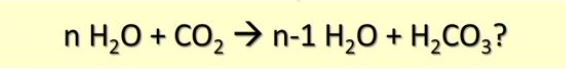
大家都知道,二氧化碳和水反应会生成碳酸,一个不可或缺的反应。那么你知道在一个碳酸分子形成过程中,有几个水分子参与么?[5][6][7]
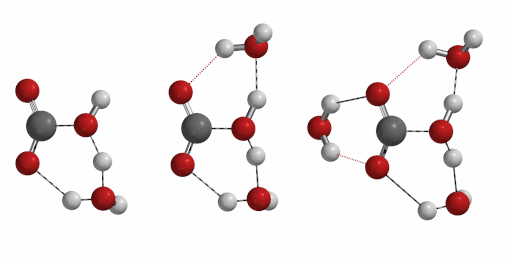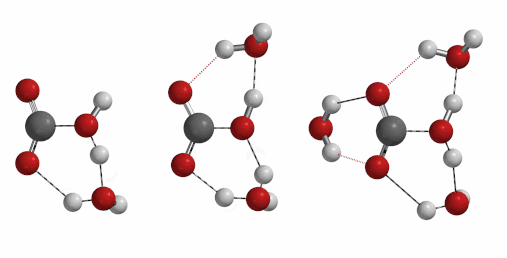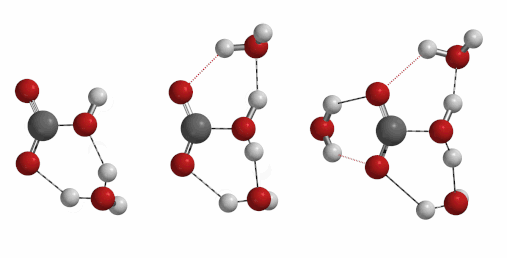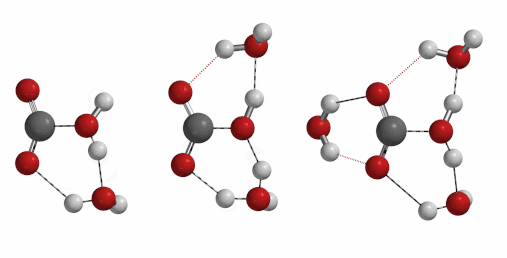
iFreq 732 cm-1 iFreq 1032 cm-1 iFreq 39 cm-1 (不相关振动)
参考文献:
[1] X.Y. Jiang, X. Hao, L.L Jing, G.C Wu, D.W. Kang, X.Y. Liu, P. Zhan, Expert Opin Drug Discov, 2019, 14, 779.
[2] G.Y. Meng, T.J. Guo, T.C. Ma, J. Zhang, Y.C. Shen, K.B. Sharpless, J.J. Dong, Nature, 2019, 574, 86.
[3] A.K. Pandiakumar, S.P. Sarma, A.G. Samuelson, Tetrahedron Lett. 2014, 55, 2917.
[4] D.C. Young, Computational Chemistry: A Practical Guide for Applying Techniques to Real-World Problems, Wiley, New York (2001).
[5] M.T. Nguyen, G. Raspoet, L.G. Vanquickenborne, J. Phys. Chem. A 1997, 101, 7379 (Molecular Orbital Model: Water self catalysis, n = 3).
[6] R.K. Lam, A.H. England, A.T. Sheardy, O. Shih, J.W. Smith, A.M. Rizzuto, D. Prendergast, R.J. Saykally, Chem. Phys. Lett., 2014, 614, 282. (First Principles Molecular Dynamic Model and X-Ray Absorption Spectroscopy: Hydration number = 3.17)
[7] S. Lindskog, Pharmacol. Ther., 1997, 74, 1. (Zn++ Catalysis, n = 1)