 选择语言
选择语言



新闻和活动

关于我们
服务和解决方案
新闻和活动
职业发展
服务平台
QM有机化学课堂
市场活动
新闻
QM有机化学课堂
我们在前面的QM小课堂学习了HOMO、LUMO概念,以及他们在有机合成中的应用。本章节我们将结合前面所学内容来设计合成路线,以及利用碳原子的核磁共振谱图来验证合成的化合物结构 [1]
现在请思考:如何从原料 (1) 快速合成目标分子 (4) 呢?原料 (1) 有多个反应位点,我们能否通过QM计算来预测序列反应的反应位点?
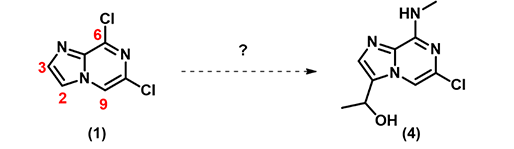
图1:以原料 (1) 合成目标分子 (4)
上述官能团的转化并不复杂,首先我们需要在2位引入卤素,芳基卤与格氏试剂交换后与醛反应得到(3) ,最后再通过SNAr反应引入胺基。
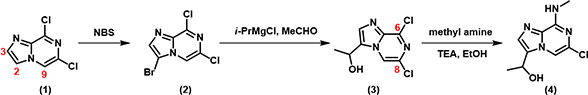
图2:目标分子 (4) 的合成路线
首先我们要解决的是芳香亲电反应的位点问题[2,3]。在第二节小课堂中,我们学习了利用HOMO来预测芳香亲电反应的位点,还有计算碳原子的核磁共振谱图,两者结合给出进一步的判断。那我们在这里再来验证一下这些方法。
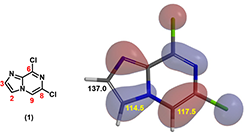
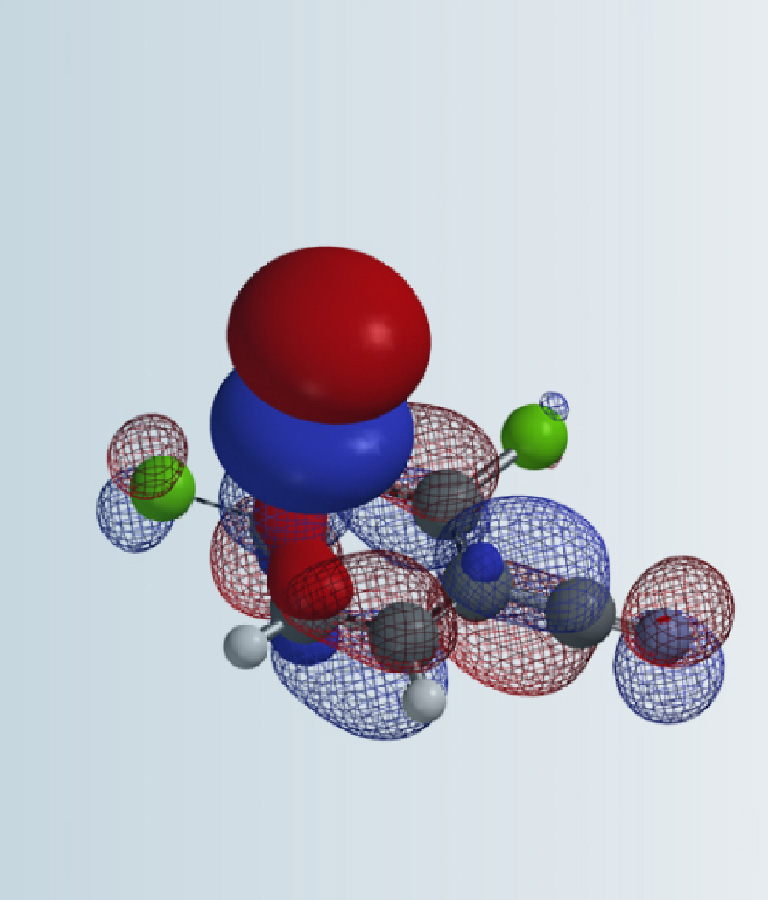
图3:原料 (1) 的HOMO及碳原子的化学位移
如图3的HOMO示意图可见,底物 (1) 中,咪唑上C3位没有HOMO lobe,C2位HOMO lobe较大;同时C2 (114.5 ppm) 与C3 (137.0 ppm) 位移值差距明显,C2位明显化学位移更低,相对更富电性。因此可以推断亲电溴化反应C2位会优先于C3位,得到C2位溴化的主要产物。
但是,小伙伴们如果仔细观察图3,会发现吡嗪上的C9位无论是HOMO lobe还是碳原子化学位移,都和C2非常相近。我们都是化学家,根据经验咪唑环是富电性环系,吡嗪环是缺电性环系,尤其是它还带着两个吸电性的氯原子,亲电取代反应毫无疑问应该发生在更富电性的咪唑上。那QM有没有办法来回答这个问题呢?
别急,我们在上节课(第十七章)介绍了通过计算亲电卤代正离子的相对能量来预测卤代反应的区域选择性。这里我们再用QM计算一下C2、C3、C9卤代正离子的平衡构象以及相对能量,下图可以很明显看出(2B) 的相对能量远低于 (2C) 以及(2A)。结合多方面QM计算数据及分析,我们预测溴化反应优先发生在C2位。
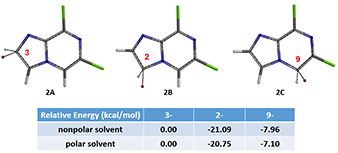
图4:C3、C2、C9位溴代正离子的平衡构象以及相对能量
中间体 (2) 中含有多个卤素原子,根据经验,Br-Mg交换的速率远大于Cl-Mg交换。如我们所预期,Br先和格氏试剂反应得到 (3) 。接下来的问题是SNAr反应的选择性,胺会优先取代哪个氯原子呢?第一节课中我们已经学习过亲核试剂会优先进攻LUMO中lobe最大的反应位点。

图5:中间体 (3) 的LUMO示意图
中间体 (3) 的LUMO图显示C6有LUMO lobe覆盖,C8 几乎没有LUMO lobe。因此亲核试剂会优先与C6位发生反应,这样的预期也和实验结果相吻合。
通过以上QM计算的综合运用,我们有效地预测了反应发生的位点,顺利拿到了最终产物。那做完了合成,我们最关心的问题之一就是,合成的究竟是不是我们预期的化合物呢?
在第十一章中,我们为大家介绍了比较碳原子的计算化学位移值和测定化学位移值来辅助解析结构异构,本章我们再次使用这个方法[4,5]。我们计算了目标化合物 (4) 的几个可能异构体,再和实验得到的数据相对比。事实证明,最终产物的碳谱实验值与目标产物的碳谱计算值最为接近,验证了我们的确获得了预期的目标产物。
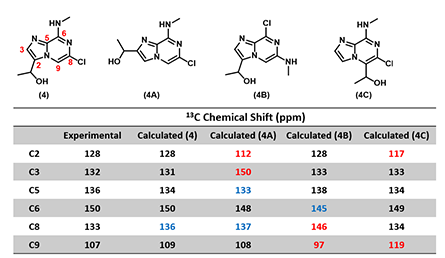
图6:终产物 (4) 的测定值与各个结构异构体QM计算碳谱化学位移值的差异(Spartan’18)
(备注: 黑色: Δ≤ 2 ppm,蓝色: 2<Δ ≤10 ppm,红色: Δ> 10 ppm。)
在本章的这一案例中,我们结合多种QM手段,分别成功预测了亲电、亲核反应的反应位点,并验证了终产物的结构。大家是不是对QM计算的应用有了更深的理解呢?
本期我们也留下一个案例给大家思考。化合物 (5) 的溴化反应经分离得到单一溴化物。大家一起来看下(5) 的13C NMR、HOMO分布,以及三个卤代正离子的相对能量,预测一下溴化会发生在哪个位点呢?

图7:化合物 (5) 的HOMO、13C NMR及C3、C4、C6位溴代正离子的平衡构象以及相对能量
本文由吴春蕊、石谷沁、王秋月、潘东、卫小文编撰。
References:
[1] J. H. Warren. A Guide to Molecular Mechanics and Quantum Chemical Calculations. Irvine, CA, USA: Wavefunction, Inc., 2003.
[2] F. A. Carey & R. J. Sundberg. Advanced Organic Chemistry Part A: Structure and Mechanisms.New York, NY, USA: Springer Science+Business Media, LLC., 2000. Pg. 551-557
[3] M. Kruszyk, M. Jessing, J. L. Kristensen, M. Jorgensen, J. Org. Chem. 2016, 81, 5128.
[4] M. Lodewyk, M. Siebert, D. Tantillo, Chem. Rev. 2012, 112, 1839.
[5] W. Hehre, P. Klunzinger, B. Deppmeier, A. Driessen, N.Uchida, M. Hashimoto, E. Fukushi, Y. Takata, J. Nat. Prod. 2019, 82, 2299